Øyedråper: Lumigan 3ml Bruk, bivirkninger og dosering. Pris i nettapotek. Generisk medisin uten resept.
Hva er Lumigan og hvordan brukes det?
Lumigan 3ml er en reseptbelagt medisin som brukes til å behandle symptomene på forhøyet intraokulært trykk forårsaket av visse typer glaukom og hyporikose i øyevippene. Lumigan kan brukes alene eller sammen med andre medisiner.
Lumigan tilhører en klasse medikamenter kalt antiglaukom, prostaglandinagonister.
Det er ikke kjent om Lumigan er trygt og effektivt hos barn.
Hva er de mulige bivirkningene av Lumigan?
Lumigan kan forårsake alvorlige bivirkninger, inkludert:
- . hevelse i øynene, . rødhet i øyet, . alvorlig ubehag i øyet, . skorpedannelse eller drenering fra øyet, . synsforandringer, og . røde, hovne eller kløende øyelokk
Få medisinsk hjelp med en gang hvis du har noen av symptomene nevnt ovenfor.
De vanligste bivirkningene av Lumigan inkluderer:
- . rødhet i øynene
Fortell legen dersom du har noen bivirkninger som plager deg eller som ikke går over.
Dette er ikke alle mulige bivirkninger av Lumigan. Spør legen din eller apoteket for mer informasjon.
Ring legen din for medisinsk råd om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
BESKRIVELSE
LUMIGAN® (bimatoprost oftalmisk løsning) 0,03 % er en syntetisk prostamidanalog med okulær hypotensiv aktivitet. Dets kjemiske navn er (Z)-7-[(1R,2R,3R,5S)-3,5-dihydroksy-2[(1E,3S)-3-hydroksy-5-fenyl-1-pentenyl]cyklopentyl]- 5-N-etylheptenamid, og dets molekylvekt er 415,58. Dens molekylformel er C25H37NO4. Dens kjemiske struktur er:
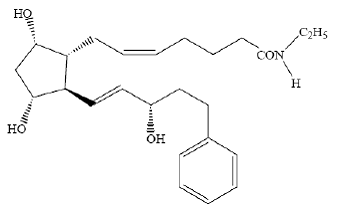
Bimatoprost er et pulver som er svært løselig i etylalkohol og metylalkohol og lett løselig i vann. LUMIGAN® 0,03 % er en klar, isotonisk, fargeløs, steril oftalmisk løsning med en osmolalitet på ca. 290 mOsmol/kg.
LUMIGAN® 0,03 % inneholder Aktiv: bimatoprost 0,3 mg/ml; Inaktive: benzalkoniumklorid 0,05 mg/ml; natriumklorid; natriumfosfat, dibasisk; sitronsyre; og renset vann. Natriumhydroksid og/eller saltsyre kan tilsettes for å justere pH. pH i løpet av holdbarheten varierer fra 6,8-7,8.
INDIKASJONER
LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 % er indisert for reduksjon av forhøyet intraokulært trykk hos pasienter med åpenvinklet glaukom eller okulær hypertensjon.
DOSERING OG ADMINISTRASJON
Anbefalt dosering er én dråpe i det/de berørte øyet en gang daglig om kvelden. LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 % bør ikke administreres mer enn én gang daglig siden det har vist seg at hyppigere administrering av prostaglandinanaloger kan redusere den intraokulære trykksenkende effekten.
Reduksjon av det intraokulære trykket starter ca. 4 timer etter første administrasjon med maksimal effekt oppnådd innen ca. 8 til 12 timer.
LUMIGAN® 0,01 % kan brukes samtidig med andre topiske oftalmiske legemidler for å senke intraokulært trykk. Hvis mer enn ett lokalt oftalmisk legemiddel brukes, bør legemidlene administreres med minst fem (5) minutters mellomrom.
HVORDAN LEVERES
Doseringsformer og styrker
Oftalmisk oppløsning som inneholder bimatoprost 0,1 mg/ml.
Oppbevaring og håndtering
LUMIGAN® (bimatoprost oftalmisk løsning) 0,01 % leveres sterilt i ugjennomsiktig hvit lavdensitets polyetylen oftalmisk dispenserflasker og spisser med turkis polystyrenhetter i følgende størrelser:
2,5 mL fyll i en 5 mL beholder - NDC 0023-3205-03 5 mL fyll i en 10 mL beholder - NDC 0023-3205-05 7,5 mL fyll i en 10 mL beholder - NDC 0023-3205-08
Oppbevaring: Oppbevares ved 2°C til 25°C (36°F til 77°F).
Etter åpning kan LUMIGAN® 0,01 % brukes frem til utløpsdatoen på flasken.
Distribuert av: Allergan USA, Inc. Madison, NJ 07940. Revidert: Mar 2022
BIVIRKNINGER
Følgende bivirkninger er beskrevet andre steder i merkingen:
- . Pigmentering inkludert blefaral pigmentering og iris hyperpigmentering [se ADVARSLER OG FORHOLDSREGLER ] . Øyevippeforandringer [se ADVARSLER OG FORHOLDSREGLER ] . Intraokulær betennelse [se ADVARSLER OG FORHOLDSREGLER ] . Makulaødem [se ADVARSLER OG FORHOLDSREGLER ] . Overfølsomhet [se KONTRAINDIKASJONER ]
Erfaring fra kliniske forsøk
Fordi kliniske studier utføres under vidt forskjellige forhold, kan bivirkningsrater observert i kliniske studier av et medikament ikke sammenlignes direkte med rater i kliniske studier av et annet medikament og gjenspeiler kanskje ikke hyppighetene observert i praksis.
en 12-måneders klinisk studie med bimatoprost oftalmiske løsninger 0,01 %, var den vanligste bivirkningen konjunktival hyperemi (31 %). Omtrent 1,6 % av pasientene avbrøt behandlingen på grunn av konjunktival hyperemi. Andre bivirkninger (rapportert hos 1 til 4 % av pasientene) med LUMIGAN® 0,01 % i denne studien inkluderte konjunktival ødem, konjunktival blødning, øyeirritasjon, øyesmerter, øyekløe, erytem i øyelokket, kløe på øyelokkene, vekst av øyevipper, hypertrikose, irritasjon på instillasjonsstedet, punktert keratitt, hudhyperpigmentering og uklart syn, redusert.
Postmarketing-erfaring
Følgende bivirkninger er identifisert under bruk etter godkjenning av LUMIGAN® 0,01 %. Fordi disse reaksjonene rapporteres frivillig fra en populasjon av usikker størrelse, er det ikke alltid mulig å pålitelig estimere frekvensen deres eller etablere en årsakssammenheng med legemiddeleksponering. Disse reaksjonene, som er valgt for inkludering enten på grunn av deres alvorlighetsgrad, hyppighet av rapportering, mulig årsakssammenheng til LUMIGAN® , eller en kombinasjon av disse faktorene inkluderer: astma-lignende symptomer, svimmelhet, tørre øyne, dyspné, øyeutflod, øyeødem, følelse av fremmedlegemer, hodepine, overfølsomhet inkludert tegn og symptomer på øyeallergi og allergisk dermatitt, hypertensjon, økt tåredannelse, periorbitale og lokkforandringer inkludert utdyping av øyelokkets sulcus og fotofobi.
NARKOTIKAHANDEL
Ingen informasjon gitt
ADVARSLER
Inkludert som en del av "FORHOLDSREGLER" Seksjon
FORHOLDSREGLER
Pigmentering
Bimatoprost oftalmisk oppløsning er rapportert å forårsake endringer i pigmentert vev. De hyppigst rapporterte endringene har vært økt pigmentering av iris, periorbitalt vev (øyelokk) og øyevipper. Pigmentering forventes å øke så lenge bimatoprost administreres. Pigmenteringsendringen skyldes økt melanininnhold i melanocyttene snarere enn en økning i antall melanocytter. Etter seponering av bimatoprost vil pigmentering av iris sannsynligvis være permanent, mens pigmentering av periorbitalvev og øyevippeforandringer er rapportert å være reversible hos noen pasienter. Pasienter som får behandling bør informeres om muligheten for økt pigmentering. Langtidseffektene av økt pigmentering er ikke kjent.
Irisfargeendring kan ikke være merkbar på flere måneder til år. Vanligvis sprer den brune pigmenteringen rundt pupillen seg konsentrisk mot periferien av iris og hele iris eller deler av iris blir mer brunaktig. Verken nevi eller fregner i iris ser ut til å være påvirket av behandlingen. Mens behandling med LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 % kan fortsettes hos pasienter som utvikler merkbart økt irispigmentering, bør disse pasientene undersøkes regelmessig.
Øyevippeforandringer
LUMIGAN® 0,01% kan gradvis endre øyevipper og vellushår i det behandlede øyet. Disse endringene inkluderer økt lengde, tykkelse og antall vipper. Vippeforandringer er vanligvis reversible ved seponering av behandlingen.
Intraokulær betennelse
Prostaglandinanaloger, inkludert bimatoprost, er rapportert å forårsake intraokulær betennelse. I tillegg, fordi disse produktene kan forverre betennelse, bør det utvises forsiktighet hos pasienter med aktiv intraokulær betennelse (f.eks. uveitt).
Makulaødem
Makulaødem, inkludert cystoid makulaødem, er rapportert under behandling med bimatoprost oftalmisk oppløsning. LUMIGAN® 0,01 % bør brukes med forsiktighet hos afake pasienter, hos pseudofake pasienter med en revet bakre linsekapsel, eller hos pasienter med kjente risikofaktorer for makulaødem.
Bakteriell keratitt
Det har vært rapporter om bakteriell keratitt assosiert med bruk av flerdosebeholdere med aktuelle oftalmiske produkter. Disse beholderne var utilsiktet blitt kontaminert av pasienter som i de fleste tilfeller hadde en samtidig hornhinnesykdom eller en forstyrrelse av den okulære epiteloverflaten.
Bruk av kontaktlinse
LUMIGAN® 0,01 % inneholder benzalkoniumklorid, som kan absorberes av og forårsake misfarging av myke kontaktlinser. Kontaktlinser bør fjernes før instillasjon av LUMIGAN® 0,01 % og kan settes inn igjen 15 minutter etter administrering.
Ikke-klinisk toksikologi
Karsinogenese, mutagenese, svekkelse av fruktbarhet
Karsinogenese
Bimatoprost var ikke kreftfremkallende hos verken mus eller rotter når det ble administrert ved oral sonde i 104 uker i doser opp til henholdsvis 2 mg/kg/dag og 1 mg/kg/dag (192 og 291 ganger estimert human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt henholdsvis én gang daglig, basert på blodets AUC-nivåer)
Mutagenese
Bimatoprost var ikke mutagent eller klastogent i Ames-testen, i muselymfomtesten eller i in vivo musemikronukleustestene.
Nedsatt fruktbarhet
Bimatoprost svekket ikke fertiliteten hos hann- eller hunnrotter opp til doser på 0,6 mg/kg/dag (minst 103 ganger anbefalt human eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig basert på blodets AUC-nivåer).
Bruk i spesifikke populasjoner
Svangerskap
Risikosammendrag
Det er ingen tilstrekkelige og godt kontrollerte studier av LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 % administrering hos gravide kvinner. Det er ingen økning i risikoen for alvorlige fødselsskader eller spontanaborter basert på erfaring med bimatoprost etter markedsføring.
I embryoføtale utviklingsstudier resulterte administrering av bimatoprost til gravide mus og rotter under organogenese i abort og tidlig fødsel ved orale doser minst 33 ganger (mus) eller 94 ganger (rotter) human eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig ( basert på blodareal under kurven [AUC] nivåer). Disse bivirkningene ble ikke observert ved 2,6 ganger (mus) og 47 ganger (rotter) human eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig (basert på blodets AUC-nivåer).
pre/postnatale utviklingsstudier resulterte administrering av bimatoprost til drektige rotter fra organogenese til slutten av ammingen i redusert drektighetslengde og føtal kroppsvekt, og økt føtal og ungdødelighet ved orale doser minst 41 ganger den humane systemiske eksponeringen for bimatoprost 0,03. % dosert bilateralt én gang daglig (basert på blodets AUC-nivåer). Ingen bivirkninger ble observert hos rotteavkom ved eksponeringer beregnet til 14 ganger human eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig (basert på blodets AUC-nivåer).
Fordi reproduksjonsstudier på dyr ikke alltid er prediktive for human respons, bør LUMIGAN® 0,01 % kun administreres under graviditet dersom den potensielle fordelen rettferdiggjør den potensielle risikoen for fosteret.
Data
Dyredata
en studie med embryoføtal utvikling av rotter ble abort observert hos gravide rotter administrert bimatoprost oralt under organogenese med 0,6 mg/kg/dag (94 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC). No Observed Adverse Effect Level (NOAEL) for abort var 0,3 mg/kg/dag (estimert til 47 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC). Ingen abnormiteter ble observert hos rottefostre ved doser opp til 0,6 mg/kg/dag.
en musestudie med embryoføtal utvikling ble abort og tidlig fødsel observert hos gravide mus administrert bimatoprost oralt under organogenese ved doser større enn eller lik 0,3 mg/kg/dag (33 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC). NOAEL for abort og tidlig fødsel var 0,1 mg/kg/dag (2,6 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC). Ingen abnormiteter ble observert hos musefoster ved doser opp til 0,6 mg/kg/dag (72 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC).
en pre/postnatal utviklingsstudie resulterte behandling av drektige rotter med bimatoprost oralt fra drektighetsdag 7 til ammedag 20 i redusert drektighetslengde, økte sene resorpsjoner, fosterdød og postnatal ungdødelighet, og redusert kroppsvekt av unger ved doser større enn eller lik 0,3 mg/kg/dag. Disse effektene ble observert ved eksponeringer på minst 41 ganger den humane systemiske eksponeringen for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC. NOAEL for postnatal utvikling og parringsytelse for avkommet var 0,1 mg/kg/dag (estimert til 14 ganger human systemisk eksponering for bimatoprost 0,03 % dosert bilateralt én gang daglig, basert på AUC).
Amming
Risikosammendrag
Det er ikke kjent om topikal okulær behandling med LUMIGAN® 0,01 % kan resultere i tilstrekkelig systemisk absorpsjon til å produsere påvisbare mengder i morsmelk. I dyrestudier har bimatoprost vist seg å være tilstede i morsmelk hos diegivende rotter i en intravenøs dose (dvs. 1 mg/kg) 970 ganger RHOD (på mg/rn 2-basis), men ingen dyredata er tilgjengelig ved klinisk relevante doser.
De utviklingsmessige og helsemessige fordelene ved amming bør vurderes sammen med mors kliniske behov for LUMIGAN® 0,01 % og eventuelle potensielle bivirkninger på det ammede barnet fra LUMIGAN® 0,01 %.
Pediatrisk bruk
Bruk hos pediatriske pasienter under 16 år anbefales ikke på grunn av potensielle sikkerhetsproblemer knyttet til økt pigmentering etter langvarig kronisk bruk.
Geriatrisk bruk
Ingen generelle kliniske forskjeller i sikkerhet eller effektivitet er observert mellom eldre og andre voksne pasienter.
OVERDOSE
Ingen informasjon er tilgjengelig om overdosering hos mennesker. Hvis overdosering med LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 % oppstår, bør behandlingen være symptomatisk.
I orale (ved sonde) studier på mus og rotter ga doser opp til 100 mg/kg/dag ingen toksisitet. Denne dosen uttrykt som mg/m2 er minst 210 ganger høyere enn tilfeldig dose av én flaske LUMIGAN® 0,01 % for et barn på 10 kg.
KONTRAINDIKASJONER
LUMIGAN® 0,01 % er kontraindisert hos pasienter med overfølsomhet overfor bimatoprost eller noen av innholdsstoffene [se BIVIRKNINGER ].
KLINISK FARMAKOLOGI
Virkningsmekanismen
Bimatoprost, en prostaglandinanalog, er en syntetisk strukturell analog av prostaglandin med okulær hypotensiv aktivitet. Den etterligner selektivt effekten av naturlig forekommende stoffer, prostamider. Bimatoprost antas å senke intraokulært trykk (IOP) hos mennesker ved å øke utstrømningen av kammervann gjennom både trabekulært nettverk og uveosklerale ruter. Forhøyet IOP utgjør en viktig risikofaktor for tap av glaukom. Jo høyere nivå av IOP, desto større er sannsynligheten for skade på synsnerven og tap av synsfelt.
Farmakokinetikk
Absorpsjon
Etter at én dråpe bimatoprost oftalmisk oppløsning 0,03 % ble administrert én gang daglig til begge øynene til 15 friske personer i to uker, toppet blodkonsentrasjonen innen 10 minutter etter dosering og var under den nedre deteksjonsgrensen (0,025 ng/ml) hos de fleste personer innen 1,5 time etter dosering. Gjennomsnittlige Cmax- og AUC0-24 timers verdier var like på dag 7 og 14 ved henholdsvis ca. 0,08 ng/ml og 0,09 ng•time/ml, noe som indikerer at steady state ble nådd i løpet av den første uken med okulær dosering. Det var ingen signifikant systemisk legemiddelakkumulering over tid.
Fordeling
Bimatoprost er moderat distribuert i kroppsvev med et steady-state distribusjonsvolum på 0,67 l/kg. I humant blod er bimatoprost hovedsakelig i plasma. Omtrent 12 % av bimatoprost forblir ubundet i humant plasma.
Eliminering
Metabolisme
Bimatoprost er den viktigste sirkulerende arten i blodet når den når den systemiske sirkulasjonen etter okulær dosering. Bimatoprost gjennomgår deretter oksidasjon, N-deetylering og glukuronidering for å danne et mangfold av metabolitter.
Utskillelse
Etter en intravenøs dose av radiomerket bimatoprost (3,12 mcg/kg) til seks friske forsøkspersoner, var den maksimale blodkonsentrasjonen av uforandret legemiddel 12,2 ng/ml og reduserte raskt med en eliminasjonshalveringstid på ca. 45 minutter. Total blodclearance av bimatoprost var 1,5 l/time/kg. Opptil 67 % av den administrerte dosen ble skilt ut i urinen mens 25 % av dosen ble gjenvunnet i avføringen.
Kliniske studier
I en 12-måneders klinisk studie av pasienter med åpenvinklet glaukom eller okulær hypertensjon med en gjennomsnittlig baseline IOP på 23,5 mmHg, var den IOP-senkende effekten av LUMIGAN® 0,01 % én gang daglig (om kvelden) opptil 7,5 mmHg.
PASIENTINFORMASJON
Potensial for pigmentering
Gi pasienter råd om potensialet for økt brun pigmentering av iris, som kan være permanent. Informer også pasientene om muligheten for mørkere øyelokkshud, som kan være reversibel etter seponering av LUMIGAN® (bimatoprost oftalmisk oppløsning) 0,01 %.
Potensial for øyevippeforandringer
Informer pasientene om muligheten for forandringer på øyevipper og vellushår i det behandlede øyet under behandling med LUMIGAN® 0,01 %. Disse endringene kan resultere i ulikhet mellom øynene i lengde, tykkelse, pigmentering, antall øyevipper eller vellushår og/eller vekstretningen for øyenvippene. Vippeforandringer er vanligvis reversible ved seponering av behandlingen.
Håndtering av beholderen
Instruer pasienter om å unngå å la tuppen av dispenseringsbeholderen komme i kontakt med øyet, omkringliggende strukturer, fingrene eller andre overflater for å unngå kontaminering av løsningen av vanlige bakterier som er kjent for å forårsake øyeinfeksjoner. Alvorlig skade på øyet og påfølgende tap av synet kan oppstå ved bruk av kontaminerte løsninger.
Når bør du søke legeråd
Informer pasienter om at dersom de utvikler en tilfeldig øyelidelse (f.eks. traumer eller infeksjon), har okulær kirurgi eller utvikler øyereaksjoner, spesielt konjunktivitt og øyelokkreaksjoner, bør de umiddelbart søke råd fra legen om fortsatt bruk av LUMIGAN® 0,01 % .
Bruk av kontaktlinse
Informer pasienter om at LUMIGAN® 0,01 % inneholder benzalkoniumklorid, som kan absorberes av myke kontaktlinser. Kontaktlinser bør fjernes før instillasjon av LUMIGAN® 0,01 % og kan settes inn igjen 15 minutter etter administrering.
Bruk sammen med andre oftalmologiske legemidler
Gi pasienter beskjed om at hvis mer enn ett lokalt oftalmisk legemiddel brukes, bør legemidlene administreres minst fem (5) minutter mellom påføringene.