Xeloda 500mg Capecitabine Bruk, bivirkninger og dosering. Pris i nettapotek. Generisk medisin uten resept.
Hva er Xeloda 500mg og hvordan brukes det?
Xeloda er et reseptbelagt legemiddel som brukes til å behandle symptomer på kreft som tykktarmskreft, tykktarmskreft og brystkreft. Xeloda kan brukes alene eller sammen med andre medisiner.
Xeloda tilhører en klasse legemidler som heter Antineoplastics, Antimetabolite.
Det er ikke kjent om Xeloda 500 mg er trygt og effektivt hos barn.
Hva er de mulige bivirkningene av Xeloda 500mg?
Xeloda kan forårsake alvorlige bivirkninger inkludert:
- . feber over 100,5 grader, . kvalme, . tap av Appetit, . spiser mye mindre enn vanlig, . oppkast (mer enn én gang i 24 timer), . alvorlig diaré (mer enn 4 ganger om dagen, eller om natten), . blemmer eller sår i munnen, . rødt eller hovent tannkjøtt, . problemer med å svelge, . smerte, ømhet, rødhet, hevelse, blemmer eller avskalling av hud på hender eller føtter, . føler meg veldig tørst eller varm, . ute av stand til å tisse, . kraftig svette, . varm og tørr hud, . brystsmerter eller trykk, . ujevne hjerteslag, . kortpustethet, . hevelse eller rask vektøkning, . smertefull eller vanskelig vannlating, . hevelse i føttene eller anklene, . føler meg sliten, . kortpustethet, . mørk urin, . leirefarget avføring, . gulfarging av hud eller øyne (gulsott), . feber eller andre influensasymptomer, . hoste, . hudsår, . blek hud, . lett blåmerker, . uvanlig blødning, . føler seg svimmel, . rask hjerterytme, . sår hals, . hevelse i ansiktet eller tungen, . brenner i øynene, og . hudsmerter som følges av rødt eller lilla utslett (spesielt ansiktet eller overkroppen) og forårsaker blemmer og avskalling
Få medisinsk hjelp med en gang hvis du har noen av symptomene nevnt ovenfor.
De vanligste bivirkningene av Xeloda 500mg inkluderer:
- . magesmerter, . forstoppelse, . urolig mage, . trøtt følelse, . mildt hudutslett, og . nummenhet eller prikking i hender eller føtter
Fortell legen dersom du har noen bivirkninger som plager deg eller som ikke går over.
Dette er ikke alle mulige bivirkninger av Xeloda. Spør legen din eller apoteket for mer informasjon.
Ring legen din for medisinsk råd om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
ADVARSEL
XELODA-WARFARIN INTERAKSJON
XELODA 500mg Warfarin Interaksjon: Pasienter som samtidig får kapecitabin og oral kumarinderivatant antikoagulantbehandling, bør få sin antikoagulantrespons (INR eller protrombintid) overvåket ofte for å justere antikoagulantdosen tilsvarende. En klinisk viktig XELODA-Warfarin legemiddelinteraksjon ble demonstrert i en klinisk farmakologisk studie [se ADVARSLER OG FORHOLDSREGLER og DRUGSINTERAKSJONER]. Endrede koagulasjonsparametere og/eller blødning, inkludert død, er rapportert hos pasienter som tar XELODA 500 mg samtidig med kumarinderivative antikoagulantia som warfarin og fenprokumon. Rapporter etter markedsføring har vist klinisk signifikant økning i protrombintid (PT) og INR hos pasienter som var stabilisert på antikoagulantia på det tidspunktet XELODA 500 mg ble introdusert. Disse hendelsene skjedde innen flere dager og opptil flere måneder etter oppstart av XELODA 500 mg-behandling og, i noen få tilfeller, innen 1 måned etter seponering av XELODA. Disse hendelsene oppsto hos pasienter med og uten levermetastaser. Alder over 60 og en kreftdiagnose disponerer pasienter uavhengig for økt risiko for koagulopati.
BESKRIVELSE
XELODA (capecitabin) er et fluoropyrimidinkarbamat med antineoplastisk aktivitet. Det er et oralt administrert systemisk prodrug av 5'-deoksy-5-fluorouridin (5'-DFUR) som omdannes til 5-fluorouracil.
Det kjemiske navnet på kapecitabin er 5'-deoksy-5-fluor-N-[(pentyloksy)karbonyl]-cytidin og har en molekylvekt på 359,35. Capecitabin har følgende strukturformel:
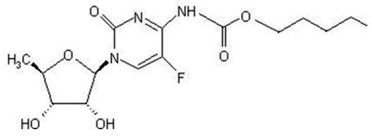
Capecitabin er et hvitt til off-white krystallinsk pulver med en vannløselighet på 26 mg/ml ved 20°C.
XELODA leveres som bikonvekse, avlange filmdrasjerte tabletter for oral administrering. Hver lys ferskenfarget tablett inneholder 150 mg capecitabin og hver ferskenfarget tablett inneholder 500 mg capecitabin. De inaktive ingrediensene i XELODA inkluderer: vannfri laktose, kroskarmellosenatrium, hydroksypropylmetylcellulose, mikrokrystallinsk cellulose, magnesiumstearat og renset vann. Fersken- eller lysferskenfilmbelegget inneholder hydroksypropylmetylcellulose, talkum, titandioksid og syntetiske gule og røde jernoksider.
INDIKASJONER
Tykktarmskreft
- . XELODA er indisert som enkeltmiddel for adjuvant behandling hos pasienter med Dukes C tykktarmskreft som har gjennomgått fullstendig reseksjon av primærtumoren når behandling med fluoropyrimidinbehandling alene er foretrukket. XELODA 500 mg var ikke dårligere enn 5fluorouracil og leukovorin (5-FU/LV) for sykdomsfri overlevelse (DFS). Leger bør vurdere resultatene av kombinasjonskemoterapistudier, som har vist forbedring i DFS og OS, når de foreskriver XELODA 500 mg enkeltmiddel i adjuvant behandling av Dukes' C tykktarmskreft. . XELODA 500mg er indisert som førstelinjebehandling av pasienter med metastatisk kolorektalt karsinom når behandling med fluoropyrimidin alene er foretrukket. Kombinasjonskjemoterapi har vist en overlevelsesfordel sammenlignet med 5-FU/LV alene. En overlevelsesfordel i forhold til 5-FU/LV er ikke vist med XELODA 500 mg monoterapi. Bruk av XELODA i stedet for 5FU/LV i kombinasjoner er ikke tilstrekkelig studert for å sikre sikkerhet eller bevaring av overlevelsesfordelen.
Brystkreft
- . XELODA i kombinasjon med docetaksel er indisert for behandling av pasienter med metastatisk brystkreft etter svikt i tidligere antracyklinholdig kjemoterapi. . XELODA 500 mg monoterapi er også indisert for behandling av pasienter med metastatisk brystkreft som er resistente mot både paklitaksel og et antracyklinholdig kjemoterapiregime eller resistente mot paklitaksel og for hvem ytterligere antracyklinbehandling ikke er indisert (f.eks. pasienter som har fått kumulative doser på 400 mg/m2 doksorubicin eller doksorubicinkvivalenter). Resistens er definert som progressiv sykdom under behandling, med eller uten initial respons, eller tilbakefall innen 6 måneder etter fullført behandling med et antracyklinholdig adjuvant regime.
DOSERING OG ADMINISTRASJON
Viktige administrasjonsinstruksjoner
XELODA tabletter skal svelges hele med vann innen 30 minutter etter et måltid. XELODA 500mg er et cellegift. Følg gjeldende spesielle håndterings- og avhendingsprosedyrer.1 Hvis XELODA 500 mg tabletter må kuttes eller knuses, bør dette gjøres av en fagperson som er opplært i sikker håndtering av cellegift ved bruk av passende utstyr og sikkerhetsprosedyrer. XELODA-dose beregnes i henhold til kroppsoverflate.
Standard startdose
Monoterapi (metastatisk kolorektal kreft, adjuvant tykktarmskreft, metastatisk brystkreft)
Den anbefalte dosen av XELODA er 1250 mg/m2 administrert oralt to ganger daglig (morgen og kveld; tilsvarende 2500 mg/m2 total daglig dose) i 2 uker etterfulgt av en 1 ukes hvileperiode gitt som 3-ukers sykluser (se ).
Adjuvant behandling hos pasienter med Dukes' C tykktarmskreft anbefales i totalt 6 måneder [dvs. XELODA 1250 mg/m2 oralt to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode, gitt som 3 ukers sykluser totalt på 8 sykluser (24 uker)].
I kombinasjon med Docetaxel (metastatisk brystkreft)
I kombinasjon med docetaksel er den anbefalte dosen av XELODA 1250 mg/m2 to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode, kombinert med docetaksel ved 75 mg/m2 som en 1-times intravenøs infusjon hver 3. uke. Premedisinering, i henhold til docetaxel-merkingen, bør startes før docetaxel-administrasjon for pasienter som får XELODA pluss docetaxel-kombinasjonen. viser den totale daglige dosen av XELODA etter kroppsoverflate og antall tabletter som skal tas ved hver dose.
Retningslinjer for dosehåndtering
Generell
XELODA-dosering må kanskje individualiseres for å optimalisere pasientbehandlingen. Pasienter bør overvåkes nøye for toksisitet og doser av XELODA 500 mg bør modifiseres etter behov for å imøtekomme individuell pasienttoleranse for behandling [se Kliniske studier ]. Toksisitet på grunn av administrering av XELODA 500 mg kan håndteres ved symptomatisk behandling, doseavbrudd og justering av XELODA-dosen. Når dosen er redusert, bør den ikke økes på et senere tidspunkt. Doser av XELODA 500 mg utelatt på grunn av toksisitet blir ikke erstattet eller gjenopprettet; i stedet bør pasienten gjenoppta de planlagte behandlingssyklusene.
Dosen av fenytoin og dosen av kumarinderivater av antikoagulantia må kanskje reduseres når begge legemidlene administreres samtidig med XELODA [se NARKOTIKAHANDEL ].
Monoterapi (metastatisk kolorektal kreft, adjuvant tykktarmskreft, metastatisk brystkreft)
XELODA dosemodifikasjonsskjema som beskrevet nedenfor (se ) anbefales for behandling av bivirkninger.
I kombinasjon med Docetaxel (metastatisk brystkreft)
Doseendringer av XELODA 500 mg for toksisitet bør gjøres i henhold til ovenfor for XELODA. Ved begynnelsen av en behandlingssyklus, hvis en behandlingsforsinkelse er indisert for enten XELODA 500 mg eller docetaxel, bør administreringen av begge legemidlene utsettes til kravene for å starte begge legemidlene på nytt er oppfylt.
Dosereduksjonsplanen for docetaksel når den brukes i kombinasjon med XELODA for behandling av metastatisk brystkreft er vist i tabell 3.
Justering av startdose i spesielle populasjoner
Nedsatt nyrefunksjon
Ingen justering av startdosen av XELODA 500 mg anbefales hos pasienter med lett nedsatt nyrefunksjon (kreatininclearance = 51 til 80 ml/min [Cockroft og Gault, som vist nedenfor]). Hos pasienter med moderat nedsatt nyrefunksjon (baseline kreatininclearance = 30 til 50 ml/min), en dosereduksjon til 75 % av XELODA-startdosen når det brukes som monoterapi eller i kombinasjon med docetaksel (fra 1250 mg/m2 til 950 mg/m2) to ganger daglig) anbefales [se Bruk i spesifikke populasjoner og KLINISK FARMAKOLOGI ]. Etterfølgende dosejustering anbefales som beskrevet i og (avhengig av regimet) hvis en pasient utvikler en grad 2 til 4 bivirkning [se ADVARSLER OG FORHOLDSREGLER ]. Anbefalingene for startdosejustering for pasienter med moderat nedsatt nyrefunksjon gjelder både XELODA 500 mg monoterapi og XELODA 500 mg i kombinasjon med docetaksel.
Cockroft og Gault-ligning:
Geriatri
Leger bør utvise forsiktighet ved å overvåke effekten av XELODA 500 mg hos eldre. Det er utilstrekkelig data tilgjengelig for å gi en doseringsanbefaling.
HVORDAN LEVERES
Doseringsformer Styrker
XELODA leveres som bikonvekse, avlange filmdrasjerte tabletter for oral administrering. Hver lys ferskenfarget tablett inneholder 150 mg capecitabin og hver ferskenfarget tablett inneholder 500 mg capecitabin.
150 mg
Farge: Lys fersken Gravering: XELODA på den ene siden og 150 på den andre 150 mg tabletter er pakket i flasker med 60 ( NDC 0004-1100-20), individuelt pakket i en kartong.
500 mg
Farge: Ferskengravering: XELODA 500 mg på den ene siden og 500 på den andre 500 mg tabletter er pakket i flasker med 120 ( NDC 0004-1101-50), individuelt pakket i en kartong.
Oppbevaring og håndtering
Oppbevares ved 25°C (77°F); turer tillatt til 15° til 30°C (59° til 86°F). [Se USP-kontrollert romtemperatur]. HOLD TETT LUKKET.
XELODA er et cellegift. Følg gjeldende spesielle håndterings- og avhendingsprosedyrer.1 Alle ubrukte preparater skal destrueres i samsvar med lokale krav, eller medikament-tilbaketagelsesprogrammer.
REFERANSER
1. "OSHA farlige stoffer." OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
Distribuert av: Genentech USA, Inc. Medlem av Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. Revidert: mai 2021
BIVIRKNINGER
Fordi kliniske studier utføres under vidt forskjellige forhold, kan bivirkningsrater observert i kliniske studier av et medikament ikke sammenlignes direkte med rater i kliniske studier av et annet medikament og gjenspeiler kanskje ikke hyppighetene observert i praksis.
Adjuvant tykktarmskreft
viser bivirkningene som forekommer hos ≥5 % av pasientene fra en fase 3-studie hos pasienter med Dukes' C tykktarmskreft som fikk minst én dose studiemedisin og hadde minst én sikkerhetsvurdering. Totalt 995 pasienter ble behandlet med 1250 mg/m2 to ganger daglig med XELODA 500 mg administrert i 2 uker etterfulgt av en 1 ukes hvileperiode, og 974 pasienter ble administrert 5-FU og leukovorin (20 mg/m2 leukovorin IV etterfulgt av 425 mg/m2 IV bolus 5FU på dag 1-5 hver 28. dag). Median behandlingsvarighet var 164 dager for capecitabinet-behandlede pasienter og 145 dager for 5-FU/LV-behandlede pasienter. Totalt 112 (11 %) og 73 (7 %) kapecitabin- og 5-FU/LV-behandlede pasienter avbrøt behandlingen på grunn av bivirkninger. Totalt 18 dødsfall på grunn av alle årsaker skjedde enten i studien eller innen 28 dager etter å ha mottatt studiemedikamentet: 8 (0,8 %) pasienter randomisert til XELODA 500 mg og 10 (1,0 %) randomisert til 5-FU/LV.
viser grad 3/4 laboratorieavvik som forekommer hos ≥1 % av pasientene fra en fase 3-studie hos pasienter med Dukes' C tykktarmskreft som fikk minst én dose studiemedisin og hadde minst én sikkerhetsvurdering.
Metastatisk tykktarmskreft
Monoterapi
viser bivirkningene som oppstår hos ≥ 5 % av pasientene fra sammenslåing av de to fase 3-studiene i førstelinjemetastatisk kolorektal kreft. Totalt 596 pasienter med metastatisk tykktarmskreft ble behandlet med 1250 mg/m2 to ganger daglig med XELODA administrert i 2 uker etterfulgt av en 1 ukes hvileperiode, og 593 pasienter ble administrert 5-FU og leukovorin i Mayo-regimet (20 mg/m2 leucovorin IV etterfulgt av 425 mg/m2 IV bolus 5-FU, på dag 1-5, hver 28. dag). I den samlede kolorektale databasen var median behandlingsvarighet 139 dager for kapecitabin-behandlede pasienter og 140 dager for 5-FU/LV-behandlede pasienter. Totalt 78 (13 %) og 63 (11 %) kapecitabin- og 5-FU/LV-behandlede pasienter avbrøt behandlingen på grunn av bivirkninger/interkurrent sykdom. Totalt 82 dødsfall på grunn av alle årsaker skjedde enten i studien eller innen 28 dager etter å ha mottatt studiemedikamentet: 50 (8,4 %) pasienter randomisert til XELODA 500 mg og 32 (5,4 %) randomisert til 5-FU/LV.
Brystkreft
I kombinasjon med Docetaxel
Følgende data er vist for kombinasjonsstudien med XELODA 500 mg og docetaksel hos pasienter med metastatisk brystkreft i og . I kombinasjonsarmen XELODA og docetaxel var behandlingen XELODA administrert oralt 1250 mg/m2 to ganger daglig som intermitterende terapi (2 ukers behandling etterfulgt av 1 uke uten behandling) i minst 6 uker og docetaxel administrert som en 1-times intravenøs infusjon kl. en dose på 75 mg/m2 den første dagen i hver 3-ukers syklus i minst 6 uker. I monoterapiarmen ble docetaxel administrert som en 1-times intravenøs infusjon i en dose på 100 mg/m2 på den første dagen av hver 3-ukers syklus i minst 6 uker. Gjennomsnittlig behandlingsvarighet var 129 dager i kombinasjonsarmen og 98 dager i monoterapiarmen. Totalt 66 pasienter (26 %) i kombinasjonsarmen og 49 (19 %) i monoterapiarmen trakk seg fra studien på grunn av bivirkninger. Andelen pasienter som trengte dosereduksjon på grunn av bivirkninger var 65 % i kombinasjonsarmen og 36 % i monoterapiarmen. Prosentandelen av pasienter som trengte behandlingsavbrudd på grunn av bivirkninger i kombinasjonsarmen var 79 %. Behandlingsavbrudd var en del av dosemodifikasjonsskjemaet for kombinasjonsterapiarmen, men ikke for docetaksel monoterapibehandlede pasienter.
Monoterapi
Følgende data er vist for studien på stadium IV brystkreftpasienter som fikk en dose på 1250 mg/m2 administrert to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode. Gjennomsnittlig behandlingsvarighet var 114 dager. Totalt 13 av 162 pasienter (8 %) avbrøt behandlingen på grunn av bivirkninger/interkurrent sykdom.
Klinisk relevante bivirkninger hos
Klinisk relevante bivirkninger rapportert hos
Monoterapi (metastatisk kolorektal kreft, adjuvant tykktarmskreft, metastatisk brystkreft)
Gastrointestinale: abdominal distensjon, dysfagi, proctalgia, ascites (0,1%), magesår (0,1%), ileus (0,3%), toksisk utvidelse av tarmen, gastroenteritt (0,1%)
Hud og subkutan.: neglelidelse (0,1 %), økt svette (0,1 %), lysfølsomhetsreaksjon (0,1 %), hudsår, kløe, strålingsgjenkallingssyndrom (0,2 %)
Generell: brystsmerter (0,2%), influensalignende sykdom, hetetokter, smerter (0,1%), heshet, irritabilitet, vansker med å gå, tørste, brystmasse, kollaps, fibrose (0,1%), blødning, ødem, sedasjon
Nevrologisk: søvnløshet, ataksi (0,5 %), tremor, dysfasi, encefalopati (0,1 %), unormal koordinasjon, dysartri, bevissthetstap (0,2 %), nedsatt balanse
Metabolisme: økt vekt, kakeksi (0,4 %), hypertriglyseridemi (0,1 %), hypokalemi, hypomagnesemi
Øye: konjunktivitt
Luftveiene: hoste (0,1 %), neseblødning (0,1 %), astma (0,2 %), hemoptyse, pustebesvær (0,1 %), dyspné
Hjerte: takykardi (0,1%), bradykardi, atrieflimmer, ventrikulære ekstrasystoler, ekstrasystoler, myokarditt (0,1%), perikardial effusjon
Infeksjoner: laryngitt (1,0%), bronkitt (0,2%), lungebetennelse (0,2%), bronkopneumoni (0,2%), keratokonjunktivitt, sepsis (0,3%), soppinfeksjoner (inkludert candidiasis) (0,2%)
Muskuloskeletal: myalgi, beinsmerter (0,1 %), leddgikt (0,1 %), muskelsvakhet
Blod og lymfe: leukopeni (0,2%), koagulasjonsforstyrrelser (0,1%), benmargsdepresjon (0,1%), idiopatisk trombocytopeni purpura (1,0%), pancytopeni (0,1%)
Vaskulær: hypotensjon (0,2%), hypertensjon (0,1%), lymfødem (0,1%), lungeemboli (0,2%), cerebrovaskulær ulykke (0,1%)
Psykiatrisk: depresjon, forvirring (0,1 %)
Nyre: nedsatt nyrefunksjon (0,6 %)
Øre: svimmelhet
Lever og galleveier: leverfibrose (0,1 %), hepatitt (0,1 %), kolestatisk hepatitt (0,1 %), unormale leverfunksjonstester
Immunforsvar: legemiddeloverfølsomhet (0,1 %)
XELODA 500mg i kombinasjon med docetaxel (metastatisk brystkreft)
Gastrointestinale: ileus (0,4 %), nekrotiserende enterokolitt (0,4 %), esophageal ulcus (0,4 %), hemorragisk diaré (0,8 %)
Nevrologisk: ataksi (0,4%), synkope (1,2%), smakstap (0,8%), polynevropati (0,4%), migrene (0,4%)
Hjerte: supraventrikulær takykardi (0,4 %)
Infeksjon: nøytropen sepsis (2,4 %), sepsis (0,4 %), bronkopneumoni (0,4 %) Blod &
Lymfe: agranulocytose (0,4 %), protrombin redusert (0,4 %)
Vaskulær: hypotensjon (1,2%), venøs flebitt og tromboflebitt (0,4%), postural hypotensjon (0,8%)
Nyre: nyresvikt (0,4 %)
Lever og galleveier: gulsott (0,4%), unormale leverfunksjonstester (0,4%), leversvikt (0,4%), leverkoma (0,4%), levertoksisitet (0,4%)
Immunforsvar: overfølsomhet (1,2 %)
Postmarketing-erfaring
Følgende bivirkninger er observert etter markedsføring: angioødem, leversvikt, tårekanalstenose, akutt nyresvikt sekundært til dehydrering inkludert fatalt utfall [se ADVARSLER OG FORHOLDSREGLER ], kutan lupus erythematosus, hornhinnesykdommer inkludert keratitt, toksisk leukoencefalopati, alvorlige hudreaksjoner som Stevens-Johnsons syndrom og toksisk epidermal nekrolyse (TEN) [se ADVARSLER OG FORHOLDSREGLER ], vedvarende eller alvorlig hånd-og-fot-syndrom kan til slutt føre til tap av fingeravtrykk [se ADVARSLER OG FORHOLDSREGLER ]
I tilfeller av eksponering for knuste XELODA 500 mg tabletter er følgende bivirkninger rapportert: øyeirritasjon og hevelse, hudutslett, diaré, parestesi, hodepine, mageirritasjon, oppkast og kvalme.
NARKOTIKAHANDEL
Legemiddel-legemiddelinteraksjoner
Antikoagulanter
Endrede koagulasjonsparametere og/eller blødninger er rapportert hos pasienter som tar XELODA 500 mg samtidig med kumarinderivative antikoagulantia som warfarin og fenprokumon [se BOKS ADVARSEL ]. Disse hendelsene skjedde innen flere dager og opptil flere måneder etter oppstart av XELODA 500 mg-behandling og, i noen få tilfeller, innen 1 måned etter seponering av XELODA. Disse hendelsene oppsto hos pasienter med og uten levermetastaser. I en legemiddelinteraksjonsstudie med enkeltdose warfarin, var det en signifikant økning i gjennomsnittlig AUC for S-warfarin [se KLINISK FARMAKOLOGI ]. Den maksimale observerte INR-verdien økte med 91 %. Denne interaksjonen skyldes sannsynligvis en hemming av cytokrom P450 2C9 av kapecitabin og/eller dets metabolitter.
Fenytoin
Nivået av fenytoin bør overvåkes nøye hos pasienter som tar XELODA 500 mg, og fenytoindosen må kanskje reduseres [se DOSERING OG ADMINISTRASJON ]. Rapporter etter markedsføring indikerer at noen pasienter som fikk XELODA 500 mg og fenytoin hadde toksisitet assosiert med forhøyede fenytoinnivåer. Formelle legemiddelinteraksjonsstudier med fenytoin er ikke utført, men interaksjonsmekanismen antas å være hemming av CYP2C9-isoenzymet av kapecitabin og/eller dets metabolitter.
Leucovorin
Konsentrasjonen av 5-fluorouracil øker og toksisiteten kan økes av leukovorin. Dødsfall fra alvorlig enterokolitt, diaré og dehydrering er rapportert hos eldre pasienter som ukentlig får leukovorin og fluorouracil.
CYP2C9 underlag
Utenom warfarin er det ikke utført noen formelle legemiddelinteraksjonsstudier mellom XELODA og andre CYP2C9-substrater. Forsiktighet bør utvises når XELODA administreres samtidig med CYP2C9-substrater.
Allopurinol
Samtidig bruk med allopurinol kan redusere konsentrasjonen av kapecitabins aktive metabolitter [se KLINISK FARMAKOLOGI ], noe som kan redusere XELODA-effekten. Unngå bruk av allopurinol under behandling med XELODA.
Interaksjon mellom narkotika og mat
Mat ble vist å redusere både hastigheten og omfanget av absorpsjon av kapecitabin [se KLINISK FARMAKOLOGI ]. I alle kliniske studier ble pasientene instruert om å administrere XELODA 500 mg innen 30 minutter etter et måltid. Det anbefales at XELODA administreres sammen med mat [se DOSERING OG ADMINISTRASJON ].
ADVARSLER
Inkludert som en del av "FORHOLDSREGLER" Seksjon
FORHOLDSREGLER
Koagulopati
Pasienter som samtidig får kapecitabin og oral antikoagulantbehandling med kumarinderivater bør få antikoagulantresponsen (INR eller protrombintid) overvåket nøye med stor hyppighet, og antikoagulantdosen bør justeres tilsvarende [se BOKS ADVARSEL og NARKOTIKAHANDEL ].
Diaré
XELODA 500 mg kan indusere diaré, noen ganger alvorlig. Pasienter med alvorlig diaré bør overvåkes nøye og gis væske- og elektrolytterstatning dersom de blir dehydrerte. Hos 875 pasienter med enten metastatisk bryst- eller tykktarmskreft som fikk XELODA monoterapi, var mediantiden til første forekomst av grad 2 til 4 diaré 34 dager (fra 1 til 369 dager). Median varighet av grad 3 til 4 diaré var 5 dager. National Cancer Institute of Canada (NCIC) grad 2 diaré er definert som en økning på 4 til 6 avføring/dag eller nattlig avføring, grad 3 diaré som en økning på 7 til 9 avføring/dag eller inkontinens og malabsorpsjon, og grad 4 diaré som en økning på ≥10 avføring/dag eller kraftig blodig diaré eller behov for parenteral støtte. Hvis grad 2, 3 eller 4 diaré oppstår, bør administreringen av XELODA avbrytes umiddelbart til diaréen går over eller reduseres i intensitet til grad 1 [se DOSERING OG ADMINISTRASJON ]. Standard antidiarébehandlinger (f.eks. loperamid) anbefales.
Nekrotiserende enterokolitt (tyflitt) er rapportert.
Kardiotoksisitet
Kardiotoksisiteten observert med XELODA 500 mg inkluderer hjerteinfarkt/iskemi, angina, dysrytmier, hjertestans, hjertesvikt, plutselig død, elektrokardiografiske endringer og kardiomyopati. Disse bivirkningene kan være mer vanlige hos pasienter med tidligere koronarsykdom.
Dihydropyrimidin-dehydrogenase-mangel
Basert på rapporter etter markedsføring har pasienter med visse homozygote eller visse sammensatte heterozygote mutasjoner i DPD-genet som resulterer i fullstendig eller nesten fullstendig fravær av DPD-aktivitet økt risiko for akutt tidlig debut av toksisitet og alvorlige, livstruende eller dødelige bivirkninger. reaksjoner forårsaket av XELODA (f.eks. mukositt, diaré, nøytropeni og nevrotoksisitet). Pasienter med delvis DPD-aktivitet kan også ha økt risiko for alvorlige, livstruende eller dødelige bivirkninger forårsaket av XELODA.
Hold tilbake eller seponer permanent XELODA 500 mg basert på klinisk vurdering av utbruddet, varigheten og alvorlighetsgraden av de observerte toksisitetene hos pasienter med tegn på akutt tidlig debut eller uvanlig alvorlig toksisitet, som kan indikere nesten fullstendig eller totalt fravær av DPD-aktivitet. Ingen XELODA-dose har vist seg å være trygg for pasienter med fullstendig fravær av DPD-aktivitet. Det er utilstrekkelig data til å anbefale en spesifikk dose hos pasienter med delvis DPD-aktivitet målt ved en spesifikk test.
Dehydrering og nyresvikt
Dehydrering er observert og kan forårsake akutt nyresvikt som kan være dødelig. Pasienter med allerede nedsatt nyrefunksjon eller som samtidig får XELODA og kjente nefrotoksiske midler har høyere risiko. Pasienter med anoreksi, asteni, kvalme, oppkast eller diaré kan raskt bli dehydrert. Overvåk pasienter når XELODA 500 mg administreres for å forhindre og korrigere dehydrering ved utbruddet. Hvis dehydrering av grad 2 (eller høyere) oppstår, bør XELODA-behandlingen avbrytes umiddelbart og dehydreringen korrigeres. Behandlingen bør ikke startes på nytt før pasienten er rehydrert og eventuelle utløsende årsaker er korrigert eller kontrollert. Doseendringer bør gjøres for den utløsende bivirkningen etter behov [se DOSERING OG ADMINISTRASJON ].
Pasienter med moderat nedsatt nyrefunksjon ved baseline trenger dosereduksjon [se DOSERING OG ADMINISTRASJON ]. Pasienter med lett og moderat nedsatt nyrefunksjon ved baseline bør overvåkes nøye for bivirkninger. Umiddelbar avbrytelse av behandlingen med påfølgende dosejusteringer anbefales hvis en pasient utvikler en grad 2 til 4 bivirkning som beskrevet i [se DOSERING OG ADMINISTRASJON , Bruk i spesifikke populasjoner , og KLINISK FARMAKOLOGI ].
Embryo-føtal toksisitet
Basert på funn fra reproduksjonsstudier på dyr og dets virkningsmekanisme, kan XELODA forårsake fosterskader når det gis til en gravid kvinne [se KLINISK FARMAKOLOGI ]. Begrensede tilgjengelige data er ikke tilstrekkelige til å informere om bruk av XELODA 500mg hos gravide kvinner. I reproduksjonsstudier på dyr forårsaket administrering av capecitabin til drektige dyr i løpet av organogeneseperioden embryodødelighet og teratogenisitet hos mus og embryodødelighet hos aper ved 0,2 og 0,6 ganger eksponeringen (AUC) hos pasienter som fikk henholdsvis anbefalt dose [se Bruk i spesifikke populasjoner ]. Gjør gravide kvinner oppmerksom på den potensielle risikoen for et foster. Informer kvinner med reproduksjonspotensial til å bruke effektiv prevensjon under behandlingen og i 6 måneder etter siste dose av XELODA [se Bruk i spesifikke populasjoner ].
Mukokutan og dermatologisk toksisitet
Alvorlige slimhinnereaksjoner, noen med dødelig utgang, som Stevens-Johnsons syndrom og toksisk epidermal nekrolyse (TEN) kan forekomme hos pasienter behandlet med XELODA [se BIVIRKNINGER ]. XELODA 500 mg bør seponeres permanent hos pasienter som opplever en alvorlig mukokutan reaksjon som muligens kan tilskrives XELODA-behandling.
Hånd-og-fot-syndrom (palmar-plantar erytrodysestesi eller kjemoterapi-indusert akral erytem) er en kutan toksisitet. Median tid til utbruddet var 79 dager (fra 11 til 360 dager) med et alvorlighetsnivå på grad 1 til 3 for pasienter som fikk XELODA 500 mg monoterapi i metastatisk setting. Grad 1 er karakterisert ved noe av følgende: nummenhet, dysestesi/parestesi, prikking, smertefri hevelse eller erytem i hender og/eller føtter og/eller ubehag som ikke forstyrrer normale aktiviteter. Grad 2 hånd-og-fot-syndrom er definert som smertefullt erytem og hevelse i hender og/eller føtter og/eller ubehag som påvirker pasientens daglige aktiviteter. Grad 3 hånd-og-fot-syndrom er definert som fuktig avskalling, sårdannelse, blemmer eller sterke smerter i hender og/eller føtter og/eller alvorlig ubehag som gjør at pasienten ikke kan arbeide eller utføre daglige aktiviteter. Vedvarende eller alvorlig hånd- og fotsyndrom (grad 2 og høyere) kan til slutt føre til tap av fingeravtrykk som kan påvirke pasientens identifikasjon. Hvis grad 2 eller 3 hånd- og fotsyndrom oppstår, bør administreringen av XELODA avbrytes inntil hendelsen forsvinner eller reduseres i intensitet til grad 1. Etter grad 3 hånd- og fotsyndrom bør påfølgende doser av XELODA 500 mg reduseres [ se DOSERING OG ADMINISTRASJON ].
Hyperbilirubinemi
Hos 875 pasienter med enten metastaserende brystkreft eller tykktarmskreft som fikk minst én dose XELODA 1250 mg/m2 to ganger daglig som monoterapi i 2 uker etterfulgt av en 1 ukes hvileperiode, oppstod grad 3 (1,5-3 x ULN) hyperbilirubinemi i 15,2 % (n=133) av pasientene og grad 4 (>3 x ULN) hyperbilirubinemi forekom hos 3,9 % (n=34) av pasientene. Av 566 pasienter som hadde levermetastaser ved baseline og 309 pasienter uten levermetastaser ved baseline, forekom grad 3 eller 4 hyperbilirubinemi hos henholdsvis 22,8 % og 12,3 %. Av de 167 pasientene med grad 3 eller 4 hyperbilirubinemi, hadde 18,6 % (n=31) også postbaseline-økninger (grad 1 til 4, uten økninger ved baseline) i alkalisk fosfatase og 27,5 % (n=46) hadde postbaseline-økninger i transaminaser kl. når som helst (ikke nødvendigvis samtidig). Flertallet av disse pasientene, 64,5 % (n=20) og 71,7 % (n=33), hadde levermetastaser ved baseline. I tillegg hadde 57,5 % (n=96) og 35,3 % (n=59) av de 167 pasientene forhøyelser (grad 1 til 4) ved både prebaseline og postbaseline i henholdsvis alkalisk fosfatase eller transaminaser. Bare 7,8 % (n=13) og 3,0 % (n=5) hadde grad 3 eller 4 økninger i alkalisk fosfatase eller transaminaser.
Hos de 596 pasientene behandlet med XELODA 500 mg som førstelinjebehandling for metastatisk tykktarmskreft, var forekomsten av grad 3 eller 4 hyperbilirubinemi lik den generelle sikkerhetsdatabasen for kliniske studier for XELODA 500 mg monoterapi. Median tid til debut for grad 3 eller 4 hyperbilirubinemi i kolorektal kreftpopulasjonen var 64 dager og median total bilirubin økte fra 8 μm/L ved baseline til 13 μm/L under behandling med XELODA. Av de 136 kolorektal kreftpasientene med grad 3 eller 4 hyperbilirubinemi, hadde 49 pasienter grad 3 eller 4 hyperbilirubinemi som siste målte verdi, hvorav 46 hadde levermetastaser ved baseline.
Hos 251 pasienter med metastatisk brystkreft som fikk en kombinasjon av XELODA 500 mg og docetaxel, oppstod hyperbilirubinemi grad 3 (1,5 til 3 x ULN) hos 7 % (n=17) og grad 4 (>3 x ULN) hyperbilirubinemi hos 2 % (n=5).
Hvis legemiddelrelaterte grad 3 til 4 økninger i bilirubin oppstår, bør administreringen av XELODA 500 mg avbrytes umiddelbart inntil hyperbilirubinemien synker til ≤3,0 X ULN [se anbefalte doseendringer under DOSERING OG ADMINISTRASJON ].
Hematologisk
Hos 875 pasienter med enten metastatisk bryst- eller tykktarmskreft som fikk en dose på 1250 mg/m2 administrert to ganger daglig som monoterapi i 2 uker etterfulgt av en 1 ukes hvileperiode, hadde 3,2 %, 1,7 % og 2,4 % av pasientene grad 3 eller 4 henholdsvis nøytropeni, trombocytopeni eller reduksjon i hemoglobin. Hos 251 pasienter med metastatisk brystkreft som fikk en dose XELODA i kombinasjon med docetaksel, hadde 68 % grad 3 eller 4 nøytropeni, 2,8 % hadde grad 3 eller 4 trombocytopeni, og 9,6 % hadde grad 3 eller 4 anemi.
Pasienter med baseline nøytrofiltall på
Geriatriske pasienter
Pasienter ≥80 år kan oppleve en større forekomst av grad 3 eller 4 bivirkninger. Hos 875 pasienter med enten metastatisk bryst- eller tykktarmskreft som fikk XELODA 500 mg monoterapi, opplevde 62 % av de 21 pasientene ≥80 år som ble behandlet med XELODA 500 mg en behandlingsrelatert grad 3 eller 4 bivirkning: diaré hos 6 (28,6 %) , kvalme hos 3 (14,3 %), hånd- og fotsyndrom hos 3 (14,3 %) og oppkast hos 2 (9,5 %) pasienter. Blant de 10 pasientene 70 år og eldre (ingen pasienter var >80 år) som ble behandlet med XELODA 500 mg i kombinasjon med docetaksel, opplevde 30 % (3 av 10) av pasientene grad 3 eller 4 diaré og stomatitt, og 40 % (4 av 10) opplevde grad 3 hånd-og-fot-syndrom.
Blant de 67 pasientene ≥60 år som får XELODA i kombinasjon med docetaxel, forekomsten av behandlingsrelaterte bivirkninger grad 3 eller 4, behandlingsrelaterte alvorlige bivirkninger, seponeringer på grunn av bivirkninger, behandlingsavbrudd på grunn av bivirkninger og behandling seponeringer innen de to første behandlingssyklusene var høyere enn i pasientgruppen
Hos 995 pasienter som fikk XELODA som adjuvant behandling for Dukes' C tykktarmskreft etter reseksjon av primærtumoren, opplevde 41 % av de 398 pasientene ≥65 år behandlet med XELODA en behandlingsrelatert grad 3 eller 4 bivirkning: hånd- og -fotsyndrom hos 75 (18,8%), diaré hos 52 (13,1%), stomatitt hos 12 (3,0%), nøytropeni/granulocytopeni hos 11 (2,8%), oppkast hos 6 (1,5%) og kvalme hos 5 (1,3%) %) pasienter. Hos pasienter ≥65 år (alle randomiserte populasjoner; capecitabin 188 pasienter, 5-FU/LV 208 pasienter) behandlet for Dukes' C tykktarmskreft etter reseksjon av primærtumoren, var fareforholdene for sykdomsfri overlevelse og total overlevelse for XELODA sammenlignet med 5-FU/LV var henholdsvis 1,01 (95 % KI 0,80 – 1,27) og 1,04 (95 % KI 0,79 – 1,37).
Leverinsuffisiens
Pasienter med mild til moderat leverdysfunksjon på grunn av levermetastaser bør overvåkes nøye når XELODA administreres. Effekten av alvorlig leverdysfunksjon på disposisjonen av XELODA er ikke kjent [se Bruk i spesifikke populasjoner og KLINISK FARMAKOLOGI ].
Kombinasjon med andre stoffer
Bruk av XELODA i kombinasjon med irinotekan er ikke tilstrekkelig undersøkt.
Informasjon om pasientveiledning
Råd pasienten til å lese den FDA-godkjente pasientmerkingen ( PASIENTINFORMASJON ).
Diaré
Informer pasienter som opplever grad 2 diaré (en økning på 4 til 6 avføringer/dag eller nattlig avføring) eller mer eller opplever alvorlig blodig diaré med sterke magesmerter og feber om å slutte å ta XELODA. Gi pasienter råd om bruk av antidiarébehandlinger (f.eks. loperamid) for å håndtere diaré [se ADVARSLER OG FORHOLDSREGLER ].
Kardiotoksisitet
Informer pasienter om risikoen for kardiotoksisitet og om å umiddelbart kontakte helsepersonell eller å oppsøke legevakten for ny debut av brystsmerter, kortpustethet, svimmelhet eller svimmelhet [se ADVARSLER OG FORHOLDSREGLER ].
Dihydropyrimidin-dehydrogenase-mangel
Råd pasienter til å varsle helsepersonell dersom de har en kjent DPD-mangel. Gi beskjed til pasienter hvis de har fullstendig eller nesten fullstendig fravær av DPD-aktivitet, de har økt risiko for akutt tidlig debut av toksisitet og alvorlige, livstruende eller dødelige bivirkninger forårsaket av XELODA (f.eks. mukositt, diaré, nøytropeni og nevrotoksisitet) [se ADVARSLER OG FORHOLDSREGLER ].
Dehydrering og nyresvikt
Instruer pasienter som opplever grad 2 eller høyere dehydrering (IV væsker indikert ADVARSLER OG FORHOLDSREGLER ].
Viktige administrasjonsinstruksjoner
Råd pasientene til å svelge XELODA 500 mg tabletter hele med vann innen 30 minutter etter et måltid. Råd pasienter og omsorgspersoner til ikke å knuse eller kutte XELODA-tabletter. Gi pasienter beskjed hvis de ikke kan svelge XELODA-tabletter hele, for å informere helsepersonell [se DOSERING OG ADMINISTRASJON ].
Overfølsomhet og angioødem
Informer pasienter om at XELODA kan forårsake alvorlige overfølsomhetsreaksjoner og angioødem. Informer pasienter som har kjent overfølsomhet overfor capecitabin eller 5-fluorouracil om å informere helsepersonell [se KONTRAINDIKASJONER ]. Instruer pasienter som utvikler overfølsomhetsreaksjoner eller slimhinnesymptomer (f.eks. urticaria, utslett, erytem, kløe eller hevelse i ansikt, lepper, tunge eller svelg som gjør det vanskelig å svelge eller puste) om å slutte å ta XELODA og umiddelbart kontakte helsepersonell eller å gå til en legevakt. [se BIVIRKNINGER ].
Kvalme
Instruer pasienter som opplever grad 2 kvalme (matinntaket betydelig redusert, men kan spise intermitterende) eller høyere om å slutte å ta XELODA 500 mg umiddelbart og kontakte helsepersonell for behandling av kvalme [se BIVIRKNINGER ].

Oppkast
Instruer pasienter som opplever oppkast av grad 2 (2 til 5 episoder i løpet av en 24-timers periode) eller mer om å slutte å ta XELODA umiddelbart og kontakte helsepersonell for behandling av oppkast [se BIVIRKNINGER ].
Hånd-og-fot-syndrom
Instruer pasienter som opplever grad 2 hånd- og fotsyndrom (smertefullt erytem og hevelse i hender og/eller føtter og/eller ubehag som påvirker pasientens daglige aktiviteter) eller høyere om å slutte å ta XELODA 500 mg umiddelbart og kontakte helsepersonell . Informer pasientene om at oppstart av symptomatisk behandling anbefales, og hånd- og fotsyndrom kan føre til tap av fingeravtrykk som kan påvirke personlig identifikasjon [se BIVIRKNINGER ].
Stomatitt
Informer pasienter som opplever grad 2 stomatitt (smertefullt erytem, ødem eller sår i munnen eller tungen, men kan spise) eller høyere om å slutte å ta XELODA umiddelbart og kontakte helsepersonell [se BIVIRKNINGER ].
Feber og nøytropeni
Informer pasienter som utvikler feber på 100,5 °F eller høyere eller andre tegn på potensiell infeksjon om å kontakte helsepersonell [se BIVIRKNINGER ].
Embryo-føtal toksisitet
Informer kvinner om reproduksjonspotensiale om den potensielle risikoen for et foster og å bruke effektiv prevensjon under behandling med XELODA 500 mg og i 6 måneder etter siste dose. Råd kvinner til å informere helsepersonell om en kjent eller mistenkt graviditet [se ADVARSLER OG FORHOLDSREGLER , Bruk i spesifikke populasjoner ].
Råd mannlige pasienter med kvinnelige partnere med reproduksjonspotensial til å bruke effektiv prevensjon under behandling med XELODA 500 mg og i 3 måneder etter siste dose [se Bruk i spesifikke populasjoner ].
Amming
Råd kvinner om ikke å amme under behandling med XELODA 500 mg og i 2 uker etter siste dose [se Bruk i spesifikke populasjoner ].
Ikke-klinisk toksikologi
Karsinogenese, mutagenese, svekkelse av fruktbarhet
Det er ikke utført tilstrekkelige studier som undersøker det karsinogene potensialet til kapecitabin. Capecitabin var ikke mutagent in vitro for bakterier (Ames-test) eller pattedyrceller (kinesisk hamster V79/HPRT-genmutasjonsanalyse). Capecitabin var klastogent in vitro til humane perifere blodlymfocytter, men ikke klastogent in vivo til benmarg hos mus (mikronukleustest). Fluorouracil forårsaker mutasjoner i bakterier og gjær. Fluorouracil forårsaker også kromosomavvik i musens mikronukleustest in vivo.
studier av fertilitet og generell reproduksjonsevne hos hunnmus, forstyrret orale capecitabindoser på 760 mg/kg/dag (ca. 2300 mg/m2/dag) brunst og forårsaket følgelig en reduksjon i fertilitet. Hos mus som ble gravide, overlevde ingen foster denne dosen. Forstyrrelsen i brunst var reversibel. Hos menn forårsaket denne dosen degenerative endringer i testiklene, inkludert reduksjon i antall spermatocytter og spermatider. I separate farmakokinetiske studier ga denne dosen hos mus 5'-DFUR AUC-verdier omtrent 0,7 ganger tilsvarende verdier hos pasienter som fikk den anbefalte daglige dosen.
Bruk i spesifikke populasjoner
Svangerskap
Risikosammendrag
Basert på funn i reproduksjonsstudier på dyr og dets virkningsmekanisme, kan XELODA 500 mg forårsake fosterskader når det administreres til en gravid kvinne [se KLINISK FARMAKOLOGI ]. Begrensede tilgjengelige humane data er ikke tilstrekkelige til å informere om den medikamentassosierte risikoen under graviditet. I reproduksjonsstudier på dyr forårsaket administrering av capecitabin til drektige dyr i perioden med organogenese embryodødelighet og teratogenisitet hos mus og embryodødelighet hos aper ved henholdsvis 0,2 og 0,6 ganger eksponeringen (AUC) hos pasienter som fikk anbefalt dose [se hhv. Data ]. Gjør gravide kvinner oppmerksom på den potensielle risikoen for et foster.
Estimert bakgrunnsrisiko for alvorlige fødselsskader og spontanabort for den angitte befolkningen er ukjent. I den generelle befolkningen i USA er den estimerte bakgrunnsrisikoen for alvorlige fødselsskader og spontanabort i klinisk anerkjente svangerskap henholdsvis 2-4 % og 15-20 %.
Data
Dyredata
Oral administrering av kapecitabin til gravide mus i løpet av organogeneseperioden ved en dose på 198 mg/kg/dag forårsaket misdannelser og embryodødelighet. I separate farmakokinetiske studier ga denne dosen hos mus 5'-DFUR AUC-verdier som var omtrent 0,2 ganger AUC-verdiene hos pasienter som fikk den anbefalte daglige dosen. Misdannelser hos mus inkluderte ganespalte, anoftalmi, mikroftalmi, oligodaktyli, polydaktyli, syndaktyli, kinky hale og utvidelse av cerebrale ventrikler. Oral administrering av kapecitabin til drektige aper i løpet av organogeneseperioden i en dose på 90 mg/kg/dag, forårsaket føtal dødelighet. Denne dosen ga 5'-DFUR AUC-verdier som var omtrent 0,6 ganger AUC-verdiene hos pasienter som fikk den anbefalte daglige dosen.
Amming
Risikosammendrag
Det er ingen informasjon om tilstedeværelsen av capecitabin i morsmelk, eller om dets effekt på melkeproduksjonen eller det ammede spedbarnet. Capecitabin-metabolitter var tilstede i melken til diegivende mus [se Data ]. På grunn av potensialet for alvorlige bivirkninger fra eksponering for kapecitabin hos spedbarn som ammes, bør kvinner rådes til ikke å amme under behandling med XELODA 500 mg og i 2 uker etter siste dose.
Data
Diegivende mus som fikk en enkelt oral dose kapecitabin utskilte betydelige mengder kapecitabin-metabolitter i melken.
Kvinner og menn med reproduktivt potensial
Graviditetstesting
Graviditetstesting anbefales for kvinner med reproduksjonspotensial før oppstart med XELODA.
Prevensjon
Kvinner
XELODA 500mg kan forårsake fosterskader når det gis til en gravid kvinne [se Svangerskap ]. Informer kvinner med reproduksjonsevne til å bruke effektiv prevensjon under behandlingen og i 6 måneder etter siste dose av XELODA.
Hanner
Basert på genetisk toksisitetsfunn, råd mannlige pasienter med kvinnelige partnere med reproduksjonspotensiale til å bruke effektiv prevensjon under behandlingen og i 3 måneder etter den siste dosen av XELODA [se Ikke-klinisk toksikologi ].
Infertilitet
Basert på dyrestudier kan XELODA svekke fertiliteten hos kvinner og menn med reproduksjonspotensial [se Ikke-klinisk toksikologi ].
Pediatrisk bruk
Sikkerhet og effektivitet av XELODA hos pediatriske pasienter er ikke fastslått. Ingen klinisk fordel ble vist i to enkeltarmsstudier med pediatriske pasienter med nydiagnostiserte hjernestammegliomer og høygradige gliomer. I begge studiene mottok pediatriske pasienter en pediatrisk undersøkelsesformulering av capecitabin samtidig med og etter fullført strålebehandling (total dose på 5580 cGy i 180 cGy-fraksjoner). Den relative biotilgjengeligheten av undersøkelsesformuleringen til XELODA 500 mg var lik.
Den første studien ble utført på 22 pediatriske pasienter (median alder 8 år, intervall 5-17 år) med nylig diagnostiserte ikke-disseminerte iboende diffuse hjernestammegliomer og høygradige gliomer. I den dosefinnende delen av studien fikk pasienter capecitabin med samtidig strålebehandling i doser fra 500 mg/m2 til 850 mg/m2 hver 12. time i opptil 9 uker. Etter en 2 ukers pause fikk pasientene 1250 mg/m2 capecitabin hver 12. time på dag 1-14 av en 21-dagers syklus i opptil 3 sykluser. Maksimal tolerert dose (MTD) av kapecitabin administrert samtidig med strålebehandling var 650 mg/m2 hver 12. time. De viktigste dosebegrensende toksisitetene var palmar-plantar erytrodysestesi og økning av alaninaminotransferase (ALT).
Den andre studien ble utført med 34 ekstra pediatriske pasienter med nylig diagnostiserte ikke-dissimerte iboende diffuse hjernestammegliomer (median alder 7 år, område 3-16 år) og 10 pediatriske pasienter som fikk MTD av capecitabin i dosefinnende studien og møtte kvalifikasjonskriteriene for denne prøven. Alle pasienter fikk 650 mg/m2 kapecitabin hver 12. time med samtidig strålebehandling i opptil 9 uker. Etter en 2 ukers pause fikk pasientene 1250 mg/m2 capecitabin hver 12. time på dag 1-14 av en 21-dagers syklus i opptil 3 sykluser.
Det var ingen forbedring i ett års progresjonsfri overlevelse og ett års total overlevelse hos pediatriske pasienter med nylig diagnostiserte indre hjernestammegliomer som fikk capecitabin i forhold til en tilsvarende populasjon av pediatriske pasienter som deltok i andre kliniske studier.
Bivirkningsprofilen til kapecitabin var i samsvar med den kjente bivirkningsprofilen hos voksne, med unntak av laboratorieavvik som forekom mer vanlig hos pediatriske pasienter. De hyppigst rapporterte laboratorieavvikene (forekomst per pasient ≥40 %) var økt ALAT (75 %), lymfocytopeni (73 %), leukopeni (73 %), hypokalemi (68 %), trombocytopeni (57 %), hypoalbuminemi (55 %). %), nøytropeni (50 %), lav hematokrit (50 %), hypokalsemi (48 %), hypofosfatemi (45 %) og hyponatremi (45 %).
Geriatrisk bruk
Leger bør være spesielt oppmerksomme på å overvåke bivirkningene av XELODA 500 mg hos eldre [se ADVARSLER OG FORHOLDSREGLER ].
Leverinsuffisiens
Utvis forsiktighet når pasienter med mild til moderat leverdysfunksjon på grunn av levermetastaser behandles med XELODA. Effekten av alvorlig leverdysfunksjon på XELODA er ikke kjent [se ADVARSLER OG FORHOLDSREGLER og KLINISK FARMAKOLOGI ].
Nyreinsuffisiens
Pasienter med moderat (kreatininclearance = 30 til 50 ml/min) og alvorlig (kreatininclearance KONTRAINDIKASJONER , ADVARSLER OG FORHOLDSREGLER , DOSERING OG ADMINISTRASJON , og KLINISK FARMAKOLOGI ].
OVERDOSE
Symptomene på akutt overdose vil omfatte kvalme, oppkast, diaré, gastrointestinal irritasjon og blødning, og benmargsdepresjon. Medisinsk behandling av overdose bør inkludere vanlige støttende medisinske intervensjoner som tar sikte på å korrigere de kliniske manifestasjonene. Selv om ingen klinisk erfaring med bruk av dialyse som behandling for XELODA 500 mg overdose er rapportert, kan dialyse være en fordel for å redusere sirkulerende konsentrasjoner av 5'-DFUR, en lavmolekylær metabolitt av moderforbindelsen.
Enkeltdoser av XELODA 500 mg var ikke dødelige for mus, rotter og aper ved doser opp til 2000 mg/kg (2,4, 4,8 og 9,6 ganger anbefalt daglig dose for mennesker på mg/m2-basis).
KONTRAINDIKASJONER
Alvorlig nedsatt nyrefunksjon
XELODA er kontraindisert hos pasienter med alvorlig nedsatt nyrefunksjon (kreatininclearance under 30 ml/min [Cockroft og Gault]) [se Bruk i spesifikke populasjoner og KLINISK FARMAKOLOGI ].
Overfølsomhet
XELODA 500mg er kontraindisert hos pasienter med kjent overfølsomhet overfor kapecitabin eller noen av dets komponenter. XELODA er kontraindisert hos pasienter som har en kjent overfølsomhet overfor 5fluorouracil.
KLINISK FARMAKOLOGI
Virkningsmekanismen
Enzymer omdanner capecitabin til 5-fluorouracil (5-FU) in vivo. Både normale celler og tumorceller metaboliserer 5-FU til 5-fluor-2'-deoksyuridinmonofosfat (FdUMP) og 5-fluoruridintrifosfat (FUTP). Disse metabolittene forårsaker celleskade ved to forskjellige mekanismer. Først binder FdUMP og folatkofaktoren, N5-10-metylentetrahydrofolat, seg til tymidylatsyntase (TS) for å danne et kovalent bundet ternært kompleks. Denne bindingen hemmer dannelsen av tymidylat fra 2'-deoksyuridylat. Tymidylat er den nødvendige forløperen til tymidintrifosfat, som er essensielt for syntesen av DNA, slik at en mangel på denne forbindelsen kan hemme celledeling. For det andre kan nukleære transkripsjonsenzymer feilaktig inkorporere FUTP i stedet for uridintrifosfat (UTP) under syntesen av RNA. Denne metabolske feilen kan forstyrre RNA-prosessering og proteinsyntese.
Farmakokinetikk
Absorpsjon
Etter oral administrering av 1255 mg/m2 to ganger daglig til kreftpasienter, nådde kapecitabin toppnivåer i blodet i løpet av ca. 1,5 timer (Tmax) med toppnivåer av 5-FU som oppsto litt senere, etter 2 timer. Mat reduserte både hastigheten og omfanget av absorpsjon av kapecitabin med gjennomsnittlig Cmax og AUC0-∞ redusert med henholdsvis 60 % og 35 %. Cmax og AUC0-∞ for 5-FU ble også redusert med mat med henholdsvis 43 % og 21 %. Mat forsinket Tmax for både foreldre og 5-FU med 1,5 timer [se ADVARSLER OG FORHOLDSREGLER , DOSERING OG ADMINISTRASJON , og Interaksjon mellom narkotika og mat ].
Farmakokinetikken til XELODA 500 mg og dets metabolitter har blitt evaluert hos ca. 200 kreftpasienter over et doseringsområde på 500 til 3500 mg/m2/dag. Over dette området var farmakokinetikken til XELODA og dets metabolitt, 5'-DFCR, doseproporsjonale og endret seg ikke over tid. Økningen i AUC for 5'-DFUR og 5-FU var imidlertid større enn proporsjonal med doseøkningen, og AUC for 5-FU var 34 % høyere på dag 14 enn på dag 1. Variasjonen mellom pasientene i Cmax og AUC for 5-FU var større enn 85 %.
Fordeling
Plasmaproteinbinding av kapecitabin og dets metabolitter er mindre enn 60 % og er ikke konsentrasjonsavhengig. Capecitabin var primært bundet til humant albumin (omtrent 35 %). XELODA 500 mg har et lavt potensial for farmakokinetiske interaksjoner relatert til plasmaproteinbinding.
Bioaktivering og metabolisme
Capecitabin metaboliseres i stor grad enzymatisk til 5-FU. I leveren hydrolyserer en 60 kDa karboksylesterase mye av forbindelsen til 5'-deoksy-5-fluorocytidin (5'-DFCR). Cytidindeaminase, et enzym som finnes i de fleste vev, inkludert svulster, konverterer deretter 5'-DFCR til 5'-DFUR. Enzymet, tymidin-fosforylase (dThdPase), hydrolyserer deretter 5'DFUR til det aktive stoffet 5-FU. Mange vev i hele kroppen uttrykker tymidin-fosforylase. Noen humane karsinomer uttrykker dette enzymet i høyere konsentrasjoner enn omkringliggende normalt vev. Etter oral administrering av XELODA 7 dager før operasjon hos pasienter med kolorektal kreft, var medianforholdet mellom 5-FU-konsentrasjon i kolorektale svulster og tilstøtende vev 2,9 (fra 0,9 til 8,0). Disse forholdstallene har ikke blitt evaluert hos brystkreftpasienter eller sammenlignet med 5-FU-infusjon.
Metabolsk vei for kapecitabin til 5-FU
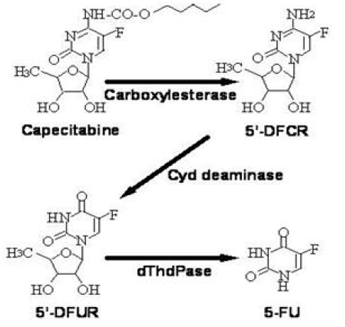
Enzymet dihydropyrimidindehydrogenase hydrogenerer 5-FU, produktet av capecitabin-metabolismen, til det mye mindre toksiske 5-fluor-5, 6-dihydro-fluorouracil (FUH2). Dihydropyrimidinase spalter pyrimidinringen for å gi 5-fluor-ureido-propionsyre (FUPA). Til slutt spalter β-ureido-propionase FUPA til α-fluor-β-alanin (FBAL) som renses i urinen.
Enzymatiske studier in vitro med humane levermikrosomer indikerte at capecitabin og dets metabolitter (5'-DFUR, 5'-DFCR, 5-FU og FBAL) ikke hemmet metabolismen av testsubstrater av cytokrom P450 isoenzymer 1A2, 2A6, 3A4, 2C19, 2D6 og 2E1.
Utskillelse
Capecitabin og dets metabolitter utskilles hovedsakelig i urin; 95,5 % av administrert capecitabin-dose gjenfinnes i urin. Fekal utskillelse er minimal (2,6%). Hovedmetabolitten som skilles ut i urinen er FBAL som representerer 57 % av den administrerte dosen. Omtrent 3 % av den administrerte dosen skilles ut i urinen som uendret legemiddel. Eliminasjonshalveringstiden for både parent kapecitabin og 5-FU var ca. 0,75 timer.
Effekt av alder, kjønn og rase på farmakokinetikken til capecitabin
En populasjonsanalyse av sammenslåtte data fra de to store kontrollerte studiene hos pasienter med metastatisk kolorektal kreft (n=505) som ble administrert XELODA 500 mg ved 1250 mg/m2 to ganger daglig, indikerte at kjønn (202 kvinner og 303 menn) og rase (455 hvite/kaukasiske pasienter, 22 svarte pasienter og 28 pasienter av annen rase) har ingen påvirkning på farmakokinetikken til 5'DFUR, 5-FU og FBAL. Alder har ingen signifikant innvirkning på farmakokinetikken til 5'-DFUR og 5-FU i området 27 til 86 år. En 20 % økning i alder resulterer i en 15 % økning i AUC for FBAL [se ADVARSLER OG FORHOLDSREGLER og DOSERING OG ADMINISTRASJON ].
Etter oral administrering av 825 mg/m2 kapecitabin to ganger daglig i 14 dager, hadde japanske pasienter (n=18) ca. 36 % lavere Cmax og 24 % lavere AUC for kapecitabin enn de kaukasiske pasientene (n=22). Japanske pasienter hadde også ca. 25 % lavere Cmax og 34 % lavere AUC for FBAL enn de kaukasiske pasientene. Den kliniske betydningen av disse forskjellene er ukjent. Ingen signifikante forskjeller forekom i eksponeringen for andre metabolitter (5'-DFCR, 5'-DFUR og 5FU).
Effekt av leverinsuffisiens
XELODA har blitt evaluert hos 13 pasienter med mild til moderat leverdysfunksjon på grunn av levermetastaser definert av en sammensatt score inkludert bilirubin, ASAT/ALT og alkalisk fosfatase etter en enkelt 1255 mg/m2 dose av XELODA. Både AUC0-∞ og Cmax for kapecitabin økte med 60 % hos pasienter med leverdysfunksjon sammenlignet med pasienter med normal leverfunksjon (n=14). AUC0-∞ og Cmax for 5-FU ble ikke påvirket. Hos pasienter med mild til moderat leverdysfunksjon på grunn av levermetastaser, bør det utvises forsiktighet når XELODA 500 mg administreres. Effekten av alvorlig leverdysfunksjon på XELODA 500 mg er ikke kjent [se ADVARSLER OG FORHOLDSREGLER og Bruk i spesielle populasjoner ].
Effekt av nyreinsuffisiens
Etter oral administrering av 1250 mg/m2 kapecitabin to ganger daglig til kreftpasienter med varierende grad av nedsatt nyrefunksjon, viste pasienter med moderat (kreatininclearance = 30 til 50 ml/min) og alvorlig (kreatininclearance 80 ml/min). Systemisk eksponering for 5'-DFUR var 42 % og 71 % høyere hos henholdsvis moderat og alvorlig nedsatt nyrefunksjon enn hos normale pasienter. Systemisk eksponering for kapecitabin var omtrent 25 % høyere hos både moderat og alvorlig nedsatt nyrefunksjon [se DOSERING OG ADMINISTRASJON , KONTRAINDIKASJONER , ADVARSLER OG FORHOLDSREGLER , og Bruk i spesielle populasjoner ].
Effekt av capecitabin på farmakokinetikken til warfarin
Hos fire pasienter med kreft økte kronisk administrering av kapecitabin (1250 mg/m2 to ganger daglig) med en enkeltdose på 20 mg warfarin gjennomsnittlig AUC for S-warfarin med 57 % og reduserte clearance med 37 %. Baseline-korrigert AUC for INR hos disse 4 pasientene økte med 2,8 ganger, og den maksimale observerte gjennomsnittlige INR-verdien ble økt med 91 % [se BOKS ADVARSEL og NARKOTIKAHANDEL ].
Effekt av antacida på farmakokinetikken til capecitabin
Da Maalox® (20 ml), et syrenøytraliserende middel som inneholder aluminiumhydroksid og magnesiumhydroksid, ble administrert umiddelbart etter XELODA (1250 mg/m2, n=12 kreftpasienter), økte AUC og Cmax med henholdsvis 16 % og 35 %. for kapecitabin og med henholdsvis 18 % og 22 % for 5'-DFCR. Ingen effekt ble observert på de tre andre hovedmetabolittene (5'-DFUR, 5-FU, FBAL) av XELODA.
Effekt av allopurinol på capecitabin
Publisert litteratur rapporterte at samtidig bruk med allopurinol kan redusere omdannelsen av kapecitabin til de aktive metabolittene, FdUMP og FUTP; den kliniske betydningen var imidlertid ikke fullstendig karakterisert.
Effekt av capecitabin på farmakokinetikken til docetaxel og omvendt
En fase 1-studie evaluerte effekten av XELODA på farmakokinetikken til docetaxel (Taxotere®) og effekten av docetaxel på farmakokinetikken til XELODA ble utført hos 26 pasienter med solide svulster. XELODA ble funnet å ikke ha noen effekt på farmakokinetikken til docetaksel (Cmax og AUC), og docetaksel har ingen effekt på farmakokinetikken til kapecitabin og 5-FU-forløperen 5'-DFUR.
Kliniske studier
Adjuvant tykktarmskreft
En multisenter randomisert, kontrollert fase 3 klinisk studie hos pasienter med Dukes' C tykktarmskreft (X-ACT) ga data om bruk av XELODA for adjuvant behandling av pasienter med tykktarmskreft. Hovedmålet med studien var å sammenligne sykdomsfri overlevelse (DFS) hos pasienter som fikk XELODA med de som fikk IV 5-FU/LV alene. I denne studien ble 1987 pasienter randomisert enten til behandling med XELODA 1250 mg/m2 oralt to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode, gitt som 3 ukers sykluser i totalt 8 sykluser (24 uker) eller IV. bolus 5-FU 425 mg/m2 og 20 mg/m2 IV leucovorin på dag 1 til 5, gitt som 4-ukers sykluser i totalt 6 sykluser (24 uker). Pasienter i studien ble pålagt å være mellom 18 og 75 år med histologisk bekreftet Dukes stadium C tykktarmskreft med minst én positiv lymfeknute og ha gjennomgått (innen 8 uker før randomisering) fullstendig reseksjon av primærtumoren uten makroskopiske eller mikroskopiske bevis på gjenværende svulst. Pasienter måtte heller ikke ha noen tidligere cytotoksisk kjemoterapi eller immunterapi (unntatt steroider), og ha en ECOG-ytelsesstatus på 0 eller 1 (KPS ≥ 70 %), ANC ≥ 1,5x109/L, blodplater ≥ 100x109/L, serumkreatinin ≤ 1,5 ULN, total bilirubin ≤ 1,5 ULN, AST/ALT ≤ 2,5 ULN og CEA innenfor normale grenser ved randomiseringstidspunktet.
Baseline-demografien for XELODA- og 5-FU/LV-pasienter er vist i tabell 10. Baseline-karakteristikkene var godt balansert mellom armene.
Alle pasienter med normal nyrefunksjon eller lett nedsatt nyrefunksjon begynte behandlingen med full startdose på 1250 mg/m2 oralt to ganger daglig. Startdosen ble redusert hos pasienter med moderat nedsatt nyrefunksjon (beregnet kreatininclearance 30 til 50 ml/min) ved baseline [se DOSERING OG ADMINISTRASJON ]. Deretter ble dosene justert for alle pasienter ved behov i henhold til toksisitet. Dosebehandling for XELODA 500 mg inkluderte dosereduksjoner, syklusforsinkelser og behandlingsavbrudd (se tabell 11).
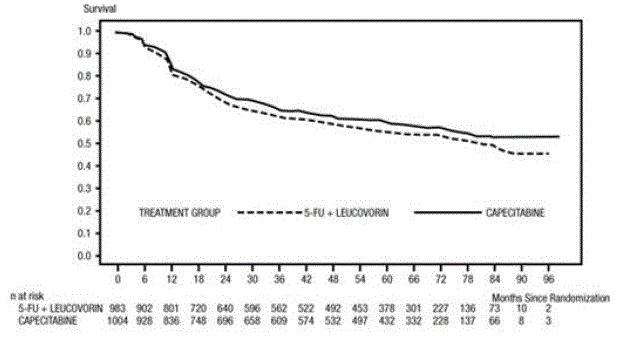
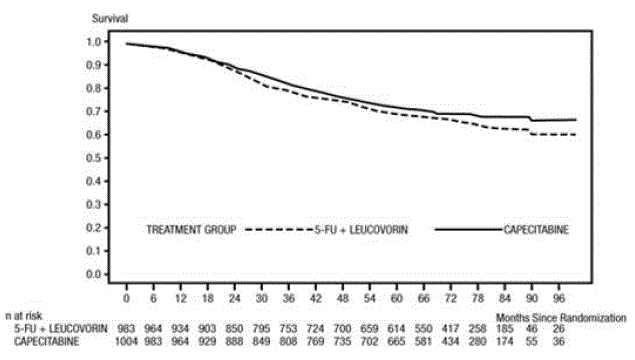
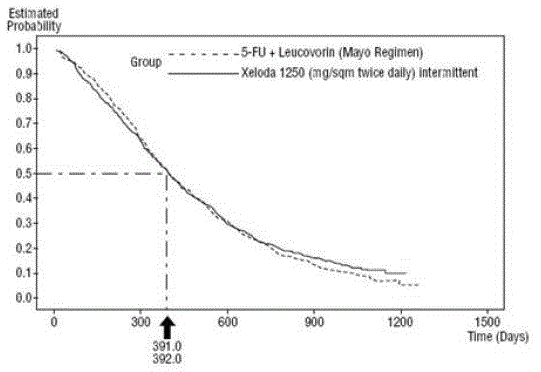
Median oppfølging ved analysetidspunktet var 83 måneder (6,9 år). Fareforholdet for DFS for XELODA 500 mg sammenlignet med 5-FU/LV var 0,88 (95 % KI 0,77 – 1,01) (se og Figur 1 ). Fordi den øvre 2-sidige 95 % konfidensgrensen for fareforhold var mindre enn 1,20, var XELODA ikke dårligere enn 5-FU/LV. Valget av non-inferiority margin på 1,20 tilsvarer retensjon av omtrent 75 % av 5-FU/LV-effekten på DFS. Hazard ratio for XELODA sammenlignet med 5-FU/LV med hensyn til total overlevelse var 0,86 (95 % KI 0,74 – 1,01). 5-års total overlevelse var 71,4 % for XELODA og 68,4 % for 5-FU/LV (se figur 2).
Figur 1 Kaplan-Meier estimater av sykdomsfri overlevelse (alle randomiserte populasjoner)a
Figur 2 Kaplan-Meier estimater for total overlevelse (alle randomiserte populasjoner)
Metastatisk tykktarmskreft
Generell
Den anbefalte dosen av XELODA 500 mg ble bestemt i en åpen, randomisert klinisk studie som undersøkte effektiviteten og sikkerheten ved kontinuerlig behandling med kapecitabin (1331 mg/m2/dag fordelt på to doser, n=39), intermitterende behandling med kapecitabin ( 2510 mg/m2/dag fordelt på to doser, n=34), og intermitterende behandling med capecitabin i kombinasjon med oral leukovorin (LV) (capecitabin 1657 mg/m2/dag fordelt på to doser, n=35; leukovorin 60 mg/dag dag) hos pasienter med avansert og/eller metastatisk kolorektalt karsinom i førstelinjemetastatisk setting. Det var ingen tilsynelatende fordel i responsrate ved å legge leucovorin til XELODA; toksisiteten ble imidlertid økt. XELODA, 1250 mg/m2 to ganger daglig i 14 dager etterfulgt av en ukes hvile, ble valgt for videre klinisk utvikling basert på den generelle sikkerhets- og effektprofilen til de tre undersøkelsene.
Monoterapi
Data fra to åpne, multisenter, randomiserte, kontrollerte kliniske studier med 1207 pasienter støtter bruken av XELODA i førstelinjebehandlingen av pasienter med metastatisk kolorektalt karsinom. De to kliniske studiene var identiske i design og ble utført i 120 sentre i forskjellige land. Studie 1 ble utført i USA, Canada, Mexico og Brasil; Studie 2 ble utført i Europa, Israel, Australia, New Zealand og Taiwan. Til sammen, i begge studiene, ble 603 pasienter randomisert til behandling med XELODA 500 mg i en dose på 1250 mg/m2 to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode og gitt som 3-ukers sykluser; 604 pasienter ble randomisert til behandling med 5-FU og leukovorin (20 mg/m2 leukovorin IV etterfulgt av 425 mg/m2 IV bolus 5-FU, på dag 1 til 5, hver 28. dag).
begge studiene ble total overlevelse, tid til progresjon og responsrate (fullstendig pluss delvis respons) vurdert. Svarene ble definert av Verdens helseorganisasjons kriterier og sendt til en blindet uavhengig revisjonskomité (IRC). Forskjeller i vurderinger mellom etterforskeren og IRC ble avstemt av sponsoren, blindet til behandlingsarmen, i henhold til en spesifisert algoritme. Overlevelse ble vurdert basert på en ikke-mindreverdighetsanalyse.
Baseline-demografien for XELODA- og 5-FU/LV-pasienter er vist i tabell 13.
Effektendepunktene for de to fase 3-studiene er vist i og
Figur 3 Kaplan-Meier-kurve for total overlevelse av sammenslåtte data (studier 1 og 2)
XELODA var overlegen 5-FU/LV for objektiv responsrate i studie 1 og studie 2. Likheten mellom XELODA 500 mg og 5-FU/LV i disse studiene ble vurdert ved å undersøke den potensielle forskjellen mellom de to behandlingene. For å sikre at XELODA 500mg har en klinisk meningsfull overlevelseseffekt, ble det utført statistiske analyser for å bestemme prosentandelen av overlevelseseffekten av 5-FU/LV som ble beholdt av XELODA. Estimatet av overlevelseseffekten av 5-FU/LV ble utledet fra en meta-analyse av ti randomiserte studier fra den publiserte litteraturen som sammenlignet 5-FU med regimer av 5-FU/LV som var lik kontrollarmene brukt i disse studiene 1 og 2. Metoden for å sammenligne behandlingene var å undersøke det verste tilfellet (95 % konfidens øvre grense) for forskjellen mellom 5-FU/LV og XELODA 500 mg, og å vise at tap på mer enn 50 % av 5- FU/LV overlevelseseffekt ble utelukket. Det ble vist at prosentandelen av overlevelseseffekten av 5-FU/LV opprettholdt var minst 61 % for studie 2 og 10 % for studie 1. Det samlede resultatet stemmer overens med en retensjon på minst 50 % av effekten på 5 -FU/LV. Det skal bemerkes at disse verdiene for bevart effekt er basert på den øvre grensen for forskjellen 5-FU/LV vs XELODA 500 mg. Disse resultatene utelukker ikke muligheten for ekte ekvivalens av XELODA 500 mg til 5-FU/LV (se og Figur 3 ).
Brystkreft
XELODA har blitt evaluert i kliniske studier i kombinasjon med docetaxel (Taxotere®) og som monoterapi.
I kombinasjon med Docetaxel
Dosen av XELODA brukt i den kliniske fase 3-studien i kombinasjon med docetaxel var basert på resultatene av en fase 1-studie, hvor en rekke doser av docetaxel administrert i 3-ukers sykluser i kombinasjon med et intermitterende regime med XELODA (14 dager) behandling, etterfulgt av en 7-dagers hvileperiode) ble evaluert. Kombinasjonsdoseregimet ble valgt basert på toleranseprofilen til 75 mg/m2 administrert i 3-ukers sykluser med docetaksel i kombinasjon med 1250 mg/m2 to ganger daglig i 14 dager med XELODA 500 mg administrert i 3-ukers sykluser. Den godkjente dosen på 100 mg/m2 docetaxel administrert i 3-ukers sykluser var kontrollarmen i fase 3-studien.
XELODA i kombinasjon med docetaxel ble vurdert i en åpen, multisenter, randomisert studie i 75 sentre i Europa, Nord-Amerika, Sør-Amerika, Asia og Australia. Totalt 511 pasienter med metastatisk brystkreft som var resistente mot, eller som gjentok seg under eller etter en antracyklinholdig behandling, eller med tilbakefall under eller tilbakevendende innen 2 år etter fullføring av en antracyklinholdig adjuvant behandling. To hundre og femtifem (255) pasienter ble randomisert til å få XELODA 1250 mg/m2 to ganger daglig i 14 dager etterfulgt av 1 uke uten behandling og docetaxel 75 mg/m2 som en 1-times intravenøs infusjon administrert i 3-ukers sykluser. I monoterapiarmen fikk 256 pasienter docetaxel 100 mg/m2 som en 1-times intravenøs infusjon administrert i 3-ukers sykluser. Pasientdemografi er gitt i tabell 16.
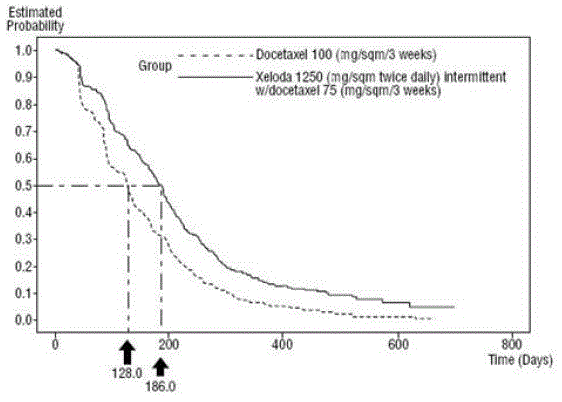
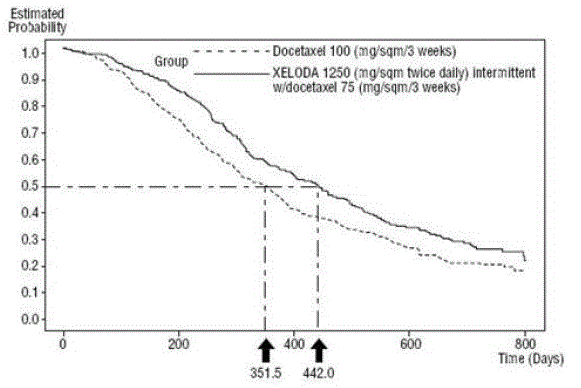
XELODA i kombinasjon med docetaxel resulterte i statistisk signifikant forbedring i tid til sykdomsprogresjon, total overlevelse og objektiv responsrate sammenlignet med monoterapi med docetaxel som vist i og Figur 5 .
Figur 4 Kaplan-Meier estimater for tid til sykdomsprogresjon XELODA og Docetaxel vs. Docetaxel
Figur 5 Kaplan-Meier estimater av overlevelse XELODA og Docetaxel vs. Docetaxel
Monoterapi
Antitumoraktiviteten til XELODA som monoterapi ble evaluert i en åpen enkeltarmsstudie utført i 24 sentre i USA og Canada. Totalt 162 pasienter med stadium IV brystkreft ble registrert. Det primære endepunktet var tumorresponsrate hos pasienter med målbar sykdom, med respons definert som en ≥50 % reduksjon i summen av produktene av de perpendikulære diametrene til bidimensjonalt målbar sykdom i minst 1 måned. XELODA ble administrert i en dose på 1255 mg/m2 to ganger daglig i 2 uker etterfulgt av en 1 ukes hvileperiode og gitt som 3 ukers sykluser. Baseline demografi og kliniske karakteristika for alle pasienter (n=162) og de med målbar sykdom (n=135) er vist i . Resistens ble definert som progressiv sykdom under behandling, med eller uten initial respons, eller tilbakefall innen 6 måneder etter fullført behandling med et antracyklinholdig adjuvant kjemoterapiregime.
Antitumorrespons for pasienter med sykdom som er resistente mot både paklitaksel og et antracyklin er vist i
For undergruppen på 43 pasienter som var dobbelt resistente, var median tid til progresjon 102 dager og median overlevelse var 255 dager. Den objektive responsraten i denne populasjonen ble støttet av en responsrate på 18,5 % (1 CR, 24 PR) i den totale populasjonen på 135 pasienter med målbar sykdom, som var mindre motstandsdyktige mot kjemoterapi (se ). Median tid til progresjon var 90 dager og median overlevelse var 306 dager.
PASIENTINFORMASJON
XELODA® (zeh-LOE-duh) (capecitabin) tabletter
Hva er den viktigste informasjonen jeg bør vite om XELODA 500mg?
XELODA 500mg kan forårsake alvorlige bivirkninger, inkludert:
- . XELODA kan samhandle med blodfortynnende medisiner, som warfarin (COUMADIN®). Å ta XELODA 500 mg sammen med disse legemidlene kan føre til endringer i hvor raskt blodpropper og kan forårsake blødninger som kan føre til døden. Dette kan skje så snart som noen dager etter at du begynner å ta XELODA 500 mg, eller senere under behandlingen, og muligens til og med innen 1 måned etter at du sluttet å ta XELODA. Risikoen kan være høyere fordi du har kreft, og hvis du er over 60 år.
- . Før du tar XELODA, fortell helsepersonell om du tar warfarin (COUMADIN) eller et annet blodfortynnende legemiddel. . Hvis du tar warfarin (COUMADIN) eller et annet blodfortynnende middel som ligner warfarin (COUMADIN) under behandling med XELODA, bør helsepersonell ta blodprøver ofte for å sjekke hvor raskt blodet ditt koagulerer under og etter at du avslutter behandlingen med XELODA. Helsepersonell kan endre dosen av blodfortynnende medisin om nødvendig.
Se "Hva er de mulige bivirkningene av XELODA 500mg?" for mer informasjon om bivirkninger.
Hva er XELODA?
XELODA er et reseptbelagt legemiddel som brukes til å behandle personer med:
- . kreft i tykktarmen som har spredt seg til lymfeknuter i området nær tykktarmen (Dukes' C-stadium), etter at de er operert. . kreft i tykktarmen eller endetarmen (kolorektal) som har spredt seg til andre deler av kroppen (metastatisk), som din første behandling av kreften på dette stadiet. . brystkreft som har spredt seg til andre deler av kroppen (metastatisk) sammen med et annet legemiddel kalt docetaxel etter at behandling med visse andre kreftmedisiner ikke har virket. . brystkreft som sprer seg til andre deler av kroppen og ikke har blitt bedre etter behandling med paklitaksel og visse andre kreftmedisiner, eller som ikke kan få mer behandling med visse kreftmedisiner.
Det er ikke kjent om XELODA 500mg er trygt og effektivt hos barn.
Ikke bruk XELODA hvis du:
- . har alvorlige nyreproblemer . er allergisk mot capecitabin, 5-fluorouracil eller noen av ingrediensene i XELODA. Se slutten av dette pakningsvedlegget for en fullstendig liste over ingredienser i XELODA.
Snakk med helsepersonell før du tar XELODA hvis du ikke er sikker på om du har noen av tilstandene nevnt ovenfor.
Før du tar XELODA, fortell helsepersonell om alle dine medisinske tilstander, inkludert hvis du:
Se "Hva er den viktigste informasjonen jeg bør vite om XELODA?"
- . har hatt hjerteproblemer . har nyre- eller leverproblemer . har blitt fortalt at du mangler enzymet DPD (dihydropyrimidindehydrogenase). . er gravid eller planlegger å bli gravid. XELODA 500 mg kan skade det ufødte barnet ditt. Helsepersonell bør ta en graviditetstest før du starter behandling med XELODA. Fortell helsepersonell umiddelbart hvis du blir gravid eller tror du kan være gravid under behandling med XELODA.
- . Kvinner som er i stand til å bli gravide bør bruke effektiv prevensjon under behandlingen og i 6 måneder etter siste dose. Snakk med helsepersonell om prevensjonsvalg som kan være riktige for deg under behandling med XELODA. . Hanner som har kvinnelige partnere som er i stand til å bli gravide, bør bruke effektiv prevensjon under behandlingen og i 3 måneder etter siste dose.
Fortell helsepersonell om alle medisinene du tar, inkludert reseptbelagte og reseptfrie medisiner, vitaminer og urtetilskudd. XELODA kan påvirke måten andre medisiner virker på, og andre medisiner kan påvirke måten XELODA virker.
Kjenn til medisinene du tar. Hold en liste over dem for å vise helsepersonell og apotek når du får en ny medisin.
Hvordan skal jeg ta XELODA 500mg?
- . Ta XELODA nøyaktig slik helsepersonell ber deg ta det. . Helsepersonell vil fortelle deg hvor mye XELODA du skal ta og når du skal ta den. . Ta XELODA 2 ganger om dagen, 1 gang om morgenen og 1 gang om kvelden. . Ta XELODA 500 mg innen 30 minutter etter et måltid. . Svelg XELODA 500 mg tabletter hele med vann. Ikke knuse eller kutte XELODA 500mg tabletter. Hvis du ikke kan svelge XELODA tabletter hele, informer helsepersonell. . Din helsepersonell kan endre dosen din, stoppe midlertidig eller permanent stoppe behandlingen med XELODA 500 mg hvis du utvikler bivirkninger. . Hvis du tar for mye XELODA, ring legen din eller gå til nærmeste legevakt umiddelbart.
Hva er de mulige bivirkningene av XELODA?
XELODA kan forårsake alvorlige bivirkninger, inkludert:
Kvalme og oppkast er vanlig med XELODA. Hvis du mister appetitten, føler deg svak og har kvalme, oppkast eller diaré, kan du raskt bli dehydrert.
Slutt å ta XELODA og ring legen din umiddelbart hvis du:
Hvis antallet hvite blodlegemer er svært lavt, har du økt risiko for infeksjon. Ring helsepersonell med en gang hvis du utvikler feber på 100,5oF eller høyere eller har andre tegn og symptomer på infeksjon.
- . Se "Hva er den viktigste informasjonen jeg bør vite om XELODA?" . Diaré. Diaré er vanlig med XELODA og kan noen ganger være alvorlig. Slutt å ta XELODA og ring legen din med en gang hvis antall avføringer du har i løpet av en dag øker med 4 eller flere avføringer enn det som er vanlig for deg eller avføring om natten. Spør helsepersonell om hvilke medisiner du kan ta for å behandle diaréen din. Hvis du har alvorlig blodig diaré med sterke magesmerter og feber, må du slutte å ta Xeloda 500 mg og ringe helsepersonell eller gå til nærmeste legevakt umiddelbart. . Hjerteproblemer. XELODA kan forårsake hjerteproblemer, inkludert: hjerteinfarkt og redusert blodtilførsel til hjertet, brystsmerter, uregelmessige hjerteslag, endringer i den elektriske aktiviteten til hjertet ditt sett på et elektrokardiogram (EKG), problemer med hjertemuskelen, hjertesvikt og plutselig død. Slutt å ta XELODA 500mg og ring helsepersonell eller gå til nærmeste legevakt umiddelbart hvis du får nye symptomer på et hjerteproblem, inkludert:
- . brystsmerter . svimmelhet . kortpustethet . svimmelhet
- . kaste opp 2 eller flere ganger på en dag. . kan bare spise eller drikke litt nå og da, eller ikke i det hele tatt på grunn av kvalme. . har diaré. Se "diaré" ovenfor.
- . XELODA 500mg kan forårsake alvorlige hudreaksjoner som kan føre til døden. Fortell helsepersonell med en gang hvis du utvikler hudutslett, blemmer og avskalling av huden din. Din helsepersonell kan fortelle deg å slutte å ta XELODA hvis du har en alvorlig hudreaksjon. Ikke ta XELODA 500 mg igjen hvis dette skjer. . XELODA 500mg kan også forårsake "hånd og fot"-syndrom. Hånd- og fotsyndrom er vanlig med XELODA 500 mg og kan føre til at du får nummenhet og endringer i følelsen i hender og føtter, eller forårsake rødhet, smerte, hevelse i hender og føtter. Slutt å ta XELODA og ring legen din med en gang hvis du har noen av disse symptomene og du ikke er i stand til å gjøre dine vanlige aktiviteter. . Hånd- og fotsyndrom kan føre til tap av fingeravtrykk som kan påvirke identifiseringen din. . du kan få sår i munnen eller på tungen når du tar XELODA. Slutt å ta XELODA 500 mg og ring legen din hvis du får smertefull rødhet, hevelse eller sår i munnen eller tungen, eller hvis du har problemer med å spise.
Personer 80 år eller eldre kan ha større sannsynlighet for å utvikle alvorlige eller alvorlige bivirkninger med XELODA.
De vanligste bivirkningene av XELODA 500mg inkluderer:
- . diaré . smerter i mageområdet (mage). . hånd- og fotsyndrom . svakhet og tretthet . kvalme . kastet opp . økte mengder nedbrytningsprodukter for røde blodlegemer (bilirubin i blodet ditt
Alvorlige allergiske reaksjoner kan oppstå med XELODA. Fortell helsepersonell dersom du noen gang har hatt en allergisk reaksjon på capecitabin eller 5-fluorouracil. Se "Ikke bruk XELODA hvis du:". Slutt å ta XELODA 500 mg og ring legen din med en gang eller gå til legevakten hvis du har noen av følgende symptomer på en alvorlig allergisk reaksjon:
- . røde kløe på huden din (elveblest) . rødhet i huden . hevelse i ansiktet, leppene, tungen eller halsen . utslett . kløe . problemer med å svelge eller puste
XELODA 500mg kan forårsake fertilitetsproblemer hos kvinner og menn. Dette kan påvirke evnen til å få barn. Snakk med helsepersonell hvis du har bekymringer om fruktbarhet.
Dette er ikke alle mulige bivirkninger av XELODA.
Ring legen din for medisinsk råd om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
Hvordan skal jeg oppbevare XELODA?
- . Oppbevar XELODA 500 mg ved romtemperatur mellom 20 °C til 25 °C (68 °F til 77 °F). . Oppbevar XELODA 500mg i en tett lukket beholder. . Spør helsepersonell eller apotek hvordan du trygt kan kaste ubrukt XELODA.
Oppbevar XELODA og alle legemidler utilgjengelig for barn.
Generell informasjon om sikker og effektiv bruk av XELODA.
Medisiner blir noen ganger foreskrevet for andre formål enn de som er oppført i et pasientinformasjonshefte. Ikke bruk XELODA for en tilstand det ikke er foreskrevet for. Ikke gi XELODA 500 mg til andre mennesker, selv om de har de samme symptomene som du har. Det kan skade dem. Du kan spørre apoteket eller helsepersonell om informasjon om XELODA 500mg som er skrevet for helsepersonell.
Hva er ingrediensene i XELODA 500mg?
Aktiv ingrediens: capecitabin
Inaktive ingredienser: vannfri laktose, kroskarmellosenatrium, hydroksypropylmetylcellulose, mikrokrystallinsk cellulose, magnesiumstearat og renset vann. Fersken- eller lysferskenfilmbelegget inneholder hydroksypropylmetylcellulose, talkum, titandioksid og syntetiske gule og røde jernoksider.
For mer informasjon, gå til http://www.gene.com/patients/medicines/xeloda 500mg eller ring 1-877-436-3683.
Denne pasientinformasjonen er godkjent av US Food and Drug Administration.