Rebetol 200mg Ribavirin Bruk, bivirkninger og dosering. Pris i nettapotek. Generisk medisin uten resept.
Hva er Rebetol 200mg og hvordan brukes det?
Rebetol er en reseptbelagt medisin som brukes til å behandle symptomene på kronisk hepatitt C. Rebetol 200mg kan brukes alene eller sammen med andre medisiner.
Rebetol tilhører en klasse medikamenter kalt hepatitt B/hepatitt C-midler; RSV-agenter.
Det er ikke kjent om Rebetol 200mg er trygt og effektivt hos barn yngre enn 3 år.
Hva er de mulige bivirkningene av Rebetol?
Rebetol kan forårsake alvorlige bivirkninger, inkludert:
- . utslett, . pustevansker, . hevelse i ansiktet, leppene, tungen eller halsen, . synsproblemer . sterke smerter i øvre del av magen sprer seg til ryggen, . kvalme, . oppkast, . diaré, . ny eller forverret hoste, . feber, . stikkende brystsmerter, . hvesing, . kortpustethet, . alvorlig depresjon, . tanker om selvskading, . tanker om å skade andre, . blek eller gulnet hud, . mørk farget urin, . forvirring, . svakhet, . frysninger, . influensalignende symptomer, . hovent tannkjøtt, . munnsår, . hudsår, . lett blåmerker, . uvanlig blødning, og . svimmelhet . Få medisinsk hjelp med en gang hvis du har noen av symptomene nevnt ovenfor. . De vanligste bivirkningene av Rebetol inkluderer: . kvalme, . oppkast, . tap av Appetit, . feber, . frysninger, . rister, . lavt antall blodceller, . anemi, . svakhet, . tretthet, . hodepine, . Muskelsmerte, . humørsvingninger, . angst, og . irritabilitet
Fortell legen dersom du har noen bivirkninger som plager deg eller som ikke går over.
Dette er ikke alle mulige bivirkninger av Rebetol. Spør legen din eller apoteket for mer informasjon.
Ring legen din for medisinsk råd om bivirkninger. Du kan rapportere bivirkninger til FDA på 1-800-FDA-1088.
ADVARSEL
RISIKO FOR ALVORLIG LIDELSER OG RIBAVIRIN-ASSOSIERTE EFFEKTER
- . REBETOL monoterapi er ikke effektivt for behandling av kronisk hepatitt C-virusinfeksjon og bør ikke brukes alene for denne indikasjonen [se ADVARSLER OG FORHOLDSREGLER]. . Den primære toksisiteten til ribavirin er hemolytisk anemi. Anemi forbundet med REBETOL 200 mg-behandling kan føre til forverring av hjertesykdom som har ført til dødelige og ikke-dødelige hjerteinfarkter. Pasienter med en historie med betydelig eller ustabil hjertesykdom skal ikke behandles med REBETOL [se DOSERING OG ADMINISTRASJON, ADVARSLER OG FORHOLDSREGLER, og BIVIRKNINGER]. . Signifikante teratogene og embryocidale effekter er påvist hos alle dyrearter som er eksponert for ribavirin. I tillegg har ribavirin en flerdose-halveringstid på 12 dager, og kan derfor vedvare i ikke-plasma-rom i så lenge som 6 måneder. Derfor er REBETOL-behandling kontraindisert hos kvinner som er gravide og hos mannlige partnere til kvinner som er gravide. Ekstrem forsiktighet må utvises for å unngå graviditet under behandlingen og i 6 måneder etter fullført behandling hos både kvinnelige pasienter og hos kvinnelige partnere til mannlige pasienter som tar REBETOL-behandling. Minst to pålitelige former for effektiv prevensjon må brukes under behandlingen og i løpet av den 6-måneders oppfølgingsperioden etter behandling [se KONTRAINDIKASJONER, ADVARSLER OG FORHOLDSREGLER, Bruk i spesifikke populasjoner, ikke-klinisk toksikologi og PASIENTINFORMASJON].
BESKRIVELSE
REBETOL (ribavirin), er en syntetisk nukleosidanalog (purinanalog). Det kjemiske navnet på ribavirin er 1-β-D-ribofuranosyl-1H-1,2,4-triazol-3-karboksamid og har følgende strukturformel (se figur 1).
Figur 1: Strukturformel
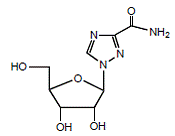
Ribavirin er et hvitt, krystallinsk pulver. Det er fritt løselig i vann og lett løselig i vannfri alkohol. Den empiriske formelen er C8H12N4O5 og molekylvekten er 244,21.
REBETOL kapsler består av et hvitt pulver i en hvit, ugjennomsiktig gelatinkapsel. Hver kapsel inneholder 200 mg ribavirin og de inaktive ingrediensene mikrokrystallinsk cellulose, laktosemonohydrat, kroskarmellosenatrium og magnesiumstearat. Kapselskallet består av gelatin, natriumlaurylsulfat, silisiumdioksid og titandioksid. Kapselen er trykket med spiselig blått farmasøytisk blekk som er laget av skjellakk, vannfri etylalkohol, isopropylalkohol, n-butylalkohol, propylenglykol, ammoniumhydroksid og FD&C Blue #2 aluminiumssjø.
REBETOL 200 mg mikstur er en klar, fargeløs til blek eller lys gul tyggegummi-smaksatt væske. Hver milliliter av oppløsningen inneholder 40 mg ribavirin og de inaktive ingrediensene sukrose, glyserin, sorbitol, propylenglykol, natriumcitrat, sitronsyre, natriumbenzoat, naturlig og kunstig smak for tyggegummi #15864 og vann.
INDIKASJONER
Kronisk hepatitt C (CHC)
REBETOL® (ribavirin) i kombinasjon med interferon alfa-2b (pegylert og ikke-pegylert) er indisert for behandling av kronisk hepatitt C (CHC) hos pasienter 3 år og eldre med kompensert leversykdom [se ADVARSLER OG FORHOLDSREGLER , og Bruk i spesifikke populasjoner ].
Følgende punkter bør vurderes når du starter REBETOL kombinasjonsbehandling med PegIntron® eller INTRON A®:
- . Disse indikasjonene er basert på oppnåelse av upåviselig HCV-RNA etter behandling i 24 eller 48 uker og opprettholdelse av en vedvarende virologisk respons (SVR) 24 uker etter siste dose. . Kombinasjonsbehandling med REBETOL/PegIntron foretrekkes fremfor REBETOL/INTRON A da denne kombinasjonen gir vesentlig bedre responsrater [se Kliniske studier ]. . Pasienter med følgende egenskaper har mindre sannsynlighet for å dra nytte av re-behandling etter mislykket behandlingsforløp: tidligere manglende respons, tidligere behandling med pegylert interferon, signifikant brodannende fibrose eller skrumplever og genotype 1-infeksjon [se Kliniske studier ]. . Ingen sikkerhets- og effektdata er tilgjengelige for behandling på mer enn ett år.
DOSERING OG ADMINISTRASJON
REBETOL-kapsler skal under ingen omstendigheter åpnes, knuses eller knuses. REBETOL 200mg bør tas sammen med mat [se KLINISK FARMAKOLOGI ]. REBETOL skal ikke brukes til pasienter med kreatininclearance mindre enn 50 ml/min.
REBETOL/PegIntron kombinasjonsterapi
Voksne pasienter
Den anbefalte dosen av PegIntron er 1,5 mcg/kg/uke subkutant i kombinasjon med 800 til 1400 mg REBETOL 200 mg kapsler oralt basert på pasientens kroppsvekt (se tabell 1). Volumet av PegIntron som skal injiseres avhenger av styrken til PegIntron og pasientens kroppsvekt, se merkingen for PegIntron for ytterligere doseringsinformasjon.
Behandlingsvarighet – Interferon alfa-naive pasienter
Behandlingsvarigheten for pasienter med genotype 1 er 48 uker. Seponering av behandlingen bør vurderes hos pasienter som ikke oppnår minst 2 log10 fall eller tap av HCV-RNA etter 12 uker, eller hvis HCV-RNA fortsatt kan påvises etter 24 ukers behandling. Pasienter med genotype 2 og 3 bør behandles i 24 uker.
Behandlingsvarighet – Re-behandling med PegIntron/REBETOL av tidligere behandlingssvikt
Behandlingsvarigheten for pasienter som tidligere mislyktes i behandlingen er 48 uker, uavhengig av HCV-genotype. Gjenbehandlede pasienter som ikke oppnår upåviselig HCV-RNA ved uke 12 av behandlingen, eller hvis HCV-RNA forblir påviselig etter 24 ukers behandling, er svært lite sannsynlig å oppnå SVR og seponering av behandlingen bør vurderes [se Kliniske studier ].
Pediatriske pasienter
Dosering for pediatriske pasienter bestemmes av kroppsoverflateareal for PegIntron og av kroppsvekt for REBETOL. Den anbefalte dosen av PegIntron er 60 mcg/m²/uke subkutant i kombinasjon med 15 mg/kg/dag av REBETOL 200 mg oralt fordelt på to doser (se tabell 2) for pediatriske pasienter i alderen 3-17 år. Pasienter som fyller 18 år mens de får PegIntron/REBETOL 200 mg, bør forbli på det pediatriske doseringsregimet. Behandlingsvarigheten for pasienter med genotype 1 er 48 uker. Pasienter med genotype 2 og 3 bør behandles i 24 uker.
REBETOL/INTRON En kombinasjonsterapi
Voksne
Behandlingsvarighet – Interferon alfa-naive pasienter
Den anbefalte dosen av INTRON A er 3 millioner IE tre ganger ukentlig subkutant. Den anbefalte dosen av REBETOL kapsler avhenger av pasientens kroppsvekt (se tabell 3). Anbefalt behandlingsvarighet for pasienter tidligere ubehandlet med interferon er 24 til 48 uker. Behandlingsvarigheten bør tilpasses pasienten avhengig av sykdomskarakteristika ved baseline, respons på terapi og tolerabilitet av regimet [se INDIKASJONER OG BRUK , BIVIRKNINGER , og Kliniske studier ]. Etter 24 ukers behandling bør virologisk respons vurderes. Seponering av behandlingen bør vurderes hos enhver pasient som ikke har oppnådd et HCV-RNA under grensen for deteksjon av analysen innen 24 uker. Det er ingen data om sikkerhet og effekt for behandling i mer enn 48 uker i tidligere ubehandlet pasientpopulasjon.
Behandlingsvarighet – Re-behandling med INTRON A/REBETOL hos tilbakefallspasienter
Hos pasienter som får tilbakefall etter ikke-pegylert interferon monoterapi, er anbefalt behandlingsvarighet 24 uker.
Pediatri
Den anbefalte dosen av REBETOL er 15 mg/kg per dag oralt (delt dose AM og PM). Se tabell 2 for pediatrisk dosering av REBETOL i kombinasjon med INTRON A. INTRON A til injeksjon etter kroppsvekt på 25 kg til 61 kg er 3 millioner IE/m² tre ganger ukentlig subkutant. Se doseringstabell for voksne for mer enn 61 kg kroppsvekt.
Anbefalt behandlingsvarighet er 48 uker for pediatriske pasienter med genotype 1. Etter 24 ukers behandling bør virologisk respons vurderes. Seponering av behandlingen bør vurderes hos enhver pasient som ikke har oppnådd et HCV-RNA under grensen for deteksjon av analysen på dette tidspunktet. Anbefalt behandlingsvarighet for pediatriske pasienter med genotype 2/3 er 24 uker.
Laboratorietester
Følgende laboratorietester anbefales for alle pasienter som behandles med REBETOL 200 mg, før behandlingsstart og deretter med jevne mellomrom.
Standard hematologiske tester - inkludert hemoglobin (forbehandling, uke 2 og uke 4 av behandlingen, og som klinisk hensiktsmessig [se ADVARSLER OG FORHOLDSREGLER ], komplette og differensielle antall hvite blodlegemer og antall blodplater.
- . Blodkjemi - leverfunksjonstester og TSH. . Graviditet - inkludert månedlig overvåking for kvinner i fertil alder. . EKG [se ADVARSLER OG FORHOLDSREGLER ].
Doseendringer
Hvis det oppstår alvorlige bivirkninger eller laboratorieavvik under kombinasjonsbehandling med REBETOL/INTRON A eller REBETOL/PegIntron, må du endre eller avbryte dosen til bivirkningen avtar eller reduseres i alvorlighetsgrad [se ADVARSLER OG FORHOLDSREGLER ]. Hvis intoleransen vedvarer etter dosejustering, bør kombinasjonsbehandlingen seponeres. Dosereduksjon av PegIntron hos voksne pasienter på REBETOL/PegIntron kombinasjonsbehandling oppnås i en to-trinns prosess fra den opprinnelige startdosen på 1,5 mcg/kg/uke, til 1 mcg/kg/uke, deretter til 0,5 mcg/kg/uke. , hvis nødvendig. Se merkingen for PegIntron for ytterligere informasjon om dosereduksjon av PegIntron.
I kombinasjonsterapistudie 2 for voksne forekom dosereduksjoner hos 42 % av pasientene som fikk PegIntron 1,5 mcg/kg og REBETOL 800 mg daglig, inkludert 57 % av pasientene som veide 60 kg eller mindre. I studie 4 hadde 16 % av forsøkspersonene en dosereduksjon av PegIntron til 1 mcg/kg i kombinasjon med REBETOL, med ytterligere 4 % som krevde den andre dosereduksjonen av PegIntron til 0,5 mcg/kg på grunn av bivirkninger [se BIVIRKNINGER ].
Dosereduksjon hos pediatriske pasienter oppnås ved å modifisere den anbefalte PegIntron-dosen i en to-trinns prosess fra den opprinnelige startdosen på 60 mcg/m²/uke, til 40 mcg/m²/uke, deretter til 20 mcg/m²/uke, hvis nødvendig (se tabell 4). I den pediatriske kombinasjonsterapistudien forekom dosereduksjoner hos 25 % av pasientene som fikk PegIntron 60 mcg/m² ukentlig og REBETOL 15 mg/kg daglig. Dosereduksjon hos pediatriske pasienter oppnås ved å endre den anbefalte REBETOL-dosen fra den opprinnelige startdosen på 15 mg/kg daglig i en to-trinns prosess til 12 mg/kg/dag, deretter til 8 mg/kg/dag, om nødvendig ( se tabell 4).
REBETOL 200 mg skal ikke brukes til pasienter med kreatininclearance mindre enn 50 ml/min. Pasienter med nedsatt nyrefunksjon og de over 50 år bør overvåkes nøye med hensyn til utvikling av anemi [se ADVARSLER OG FORHOLDSREGLER , Bruk i spesifikke populasjoner , og KLINISK FARMAKOLOGI ].
REBETOL 200 mg bør administreres med forsiktighet til pasienter med allerede eksisterende hjertesykdom. Pasienter bør vurderes før behandlingsstart og bør overvåkes hensiktsmessig under behandlingen. Hvis det er noen forverring av kardiovaskulær status, bør behandlingen avbrytes [se ADVARSLER OG FORHOLDSREGLER ].
For pasienter med stabil kardiovaskulær sykdom i anamnesen, er en permanent dosereduksjon nødvendig hvis hemoglobinet reduseres med mer enn eller lik 2 g/dL i løpet av en 4-ukers periode. Hvis hemoglobinet forblir mindre enn 12 g/dL etter 4 uker med redusert dose, bør pasienten i tillegg avbryte kombinasjonsbehandlingen for disse hjertepasientene.
Det anbefales at en pasient hvis hemoglobinnivå faller under 10 g/dL får sin REBETOL-dose endret eller seponert i henhold til tabell 4 [se ADVARSLER OG FORHOLDSREGLER ].
Se merkingen for INTRON A eller PegIntron for ytterligere informasjon om hvordan du reduserer en INTRON A- eller PegIntron-dose.
Seponering av dosering
Voksne
Hos HCV genotype 1, interferon-alfa-naive pasienter som får PegIntron i kombinasjon med ribavirin, anbefales seponering av behandlingen hvis det ikke er minst 2 log10 dråper eller tap av HCV-RNA ved 12 ukers behandling, eller hvis HCV-RNA nivåene forblir påviselige etter 24 ukers behandling. Uavhengig av genotype er det høyst usannsynlig at tidligere behandlede pasienter som har påvisbart HCV-RNA ved uke 12 eller 24 oppnår SVR, og seponering av behandlingen bør vurderes.
Pediatri (3-17 år)
Det anbefales at pasienter som får PegIntron/REBETOL-kombinasjon (unntatt HCV genotype 2 og 3) seponeres fra behandlingen etter 12 uker hvis behandlingen Uke 12 HCV-RNA falt mindre enn 2 log10 sammenlignet med en forbehandling eller ved 24 uker hvis de har påvisbar HCV-RNA ved behandling uke 24.
HVORDAN LEVERES
Doseringsformer og styrker
REBETOL 200mg Kapsler 200 mg
REBETOL 200 mg oral oppløsning 40 mg per ml
Oppbevaring og håndtering
REBETOL 200 mg kapsler er hvite, ugjennomsiktige kapsler med REBETOL, 200 mg, og Schering Corporation-logoen påtrykt på kapselskallet; kapslene er pakket i en flaske som inneholder 56 kapsler ( NDC 0085-1351-05), 70 kapsler ( NDC 0085-1385-07), og 84 kapsler ( NDC 0085-1194-03).
REBETOL 200mg mikstur 40 mg per ml er en klar, fargeløs til blek eller lys gul tyggegummi-smaksatt væske og den er pakket i 4-oz gule glassflasker (100 ml/flaske) med barnesikre lukkinger ( NDC 0085-1318-01).
Flasken med REBETOL kapsler skal oppbevares ved 25°C (77°F); utflukter tillatt til 15-30°C (59-86°F) [se USP kontrollert romtemperatur ].
REBETOL 200mg mikstur bør oppbevares mellom 2-8°C (36-46°F) eller ved 25°C (77°F); utflukter tillatt til 15-30°C (59-86°F) [se USP kontrollert romtemperatur ].
REBETOL 200mg oral løsning produsert for: Merck Sharp & Dohme Corp., et datterselskap av Whitehouse Station, NJ 08889, USA. Produsert av: Schering-Plough Canada, Inc., Pointe Claire, Quebec, Canada REBETOL Kapsler produsert av: Merck Sharp & Dohme Corp., et datterselskap av Whitehouse Station, NJ 08889, USA. Revidert: desember 2014.
BIVIRKNINGER
Kliniske studier med REBETOL i kombinasjon med PegIntron eller INTRON A har blitt utført på over 7800 forsøkspersoner fra 3 til 76 år.
Den primære toksisiteten til ribavirin er hemolytisk anemi. Reduksjoner i hemoglobinnivåer skjedde i løpet av de første 1 til 2 ukene av oral behandling. Hjerte- og lungereaksjoner assosiert med anemi forekom hos omtrent 10 % av pasientene [se ADVARSLER OG FORHOLDSREGLER ].
Mer enn 96 % av alle forsøkspersoner i kliniske studier opplevde en eller flere bivirkninger. De vanligst rapporterte bivirkningene hos voksne personer som fikk PegIntron eller INTRON A i kombinasjon med REBETOL 200 mg var betennelse/reaksjon på injeksjonsstedet, tretthet/asteni, hodepine, rigor, feber, kvalme, myalgi og angst/emosjonell labilitet/irritabilitet. De vanligste bivirkningene hos pediatriske personer i alderen 3 år og eldre som fikk REBETOL 200 mg i kombinasjon med PegIntron eller INTRON A var feber, hodepine, nøytropeni, tretthet, anoreksi, erytem på injeksjonsstedet og oppkast.
Bivirkningsdelen viser til følgende kliniske studier:
- . REBETOL/PegIntron Kombinasjonsterapiforsøk:
- . Klinisk studie 1 – evaluert PegIntron monoterapi (ikke nærmere beskrevet i denne etiketten; se merking for PegIntron for informasjon om denne studien). . Studie 2 – evaluert REBETOL 800 mg/dag flat dose i kombinasjon med 1,5 mcg/kg/uke PegIntron eller med INTRON A. . Studie 3 – evaluert PegIntron/vektbasert REBETOL 200mg i kombinasjon med PegIntron/flat dose REBETOL 200mg-regime. . Studie 4 – sammenlignet to doser PegIntron (1,5 mcg/kg/uke og 1 mcg/kg/uke) i kombinasjon med REBETOL og en tredje behandlingsgruppe som fikk Pegasys® (180 mcg/uke)/Copegus® (1000-1200 mg/dag) ). . Studie 5 – evaluerte PegIntron (1,5 mcg/kg/uke) i kombinasjon med vektbasert REBETOL hos tidligere behandlingssvikt.
Alvorlige bivirkninger har forekommet hos omtrent 12 % av pasientene i kliniske studier med PegIntron med eller uten REBETOL [se ADVARSEL i boks , ADVARSLER OG FORHOLDSREGLER ]. De vanligste alvorlige hendelsene som oppsto hos personer behandlet med PegIntron og REBETOL 200 mg var depresjon og selvmordstanker [se ADVARSLER OG FORHOLDSREGLER ], som hver forekommer med en frekvens på mindre enn 1 %. Selvmordstanker eller selvmordsforsøk forekom hyppigere blant pediatriske pasienter, først og fremst ungdom, sammenlignet med voksne pasienter (2,4 % versus 1 %) under behandling og oppfølging utenfor behandlingen [se ADVARSLER OG FORHOLDSREGLER ]. Den vanligste dødelige reaksjonen som oppstod hos personer behandlet med PegIntron og REBETOL 200 mg var hjertestans, selvmordstanker og selvmordsforsøk [se ADVARSLER OG FORHOLDSREGLER ], alle forekommer hos mindre enn 1 % av forsøkspersonene.
Fordi kliniske studier utføres under vidt forskjellige forhold, kan bivirkningsrater observert i kliniske studier av et legemiddel ikke sammenlignes direkte med rater i kliniske studier av et annet legemiddel, og gjenspeiler kanskje ikke ratene observert i klinisk praksis.
Erfaring fra kliniske studier – REBETOL/PegIntron-kombinasjonsterapi
Voksne emner
Bivirkninger som oppsto i den kliniske studien med mer enn 5 % forekomst er gitt av behandlingsgruppen fra REBETOL/PegIntron-kombinasjonsterapien (studie 2) i tabell 5.
Tabell 6 oppsummerer de behandlingsrelaterte bivirkningene i studie 4 som oppsto med en forekomst på mer enn eller lik 10 %.
Forekomsten av alvorlige bivirkninger var sammenlignbar i alle studier. I studie 3 var det en lignende forekomst av alvorlige bivirkninger rapportert for den vektbaserte REBETOL-gruppen (12 %) og for flatdose-REBETOL-regimet. I studie 2 var forekomsten av alvorlige bivirkninger 17 % i PegIntron/REBETOL-gruppene sammenlignet med 14 % i INTRON A/REBETOL-gruppen.
mange, men ikke alle tilfeller, forsvant bivirkningene etter dosereduksjon eller seponering av behandlingen. Noen forsøkspersoner opplevde pågående eller nye alvorlige bivirkninger i løpet av den 6-måneders oppfølgingsperioden. I studie 2 fortsatte mange forsøkspersoner å oppleve bivirkninger flere måneder etter seponering av behandlingen. Ved slutten av den 6-måneders oppfølgingsperioden var forekomsten av pågående bivirkninger etter kroppsklasse i gruppen PegIntron 1,5/REBETOL 200 mg 33 % (psykiatrisk), 20 % (muskuloskeletal) og 10 % (for endokrine og for GI). Hos omtrent 10 til 15 % av forsøkspersonene hadde ikke vekttap, tretthet og hodepine gått over.
Det har vært 31 pasientdødsfall som skjedde under behandling eller under oppfølging i disse kliniske studiene. I studie 1 var det 1 selvmord hos en person som fikk PegIntron monoterapi og 2 dødsfall blant personer som fikk INTRON A monoterapi (1 drap/selvmord og 1 plutselig død). I studie 2 var det 1 selvmord hos en person som fikk PegIntron/REBETOL 200 mg kombinasjonsbehandling; og 1 forsøksperson død i INTRON A/REBETOL 200mg-gruppen (motorvognulykke). I studie 3 var det 14 dødsfall, hvorav 2 var sannsynlige selvmord og 1 var et uforklarlig dødsfall hos en person med en relevant sykehistorie med depresjon. I studie 4 var det 12 dødsfall, hvorav 6 skjedde hos personer som fikk PegIntron/REBETOL kombinasjonsbehandling, 5 i PegIntron 1,5 mcg/REBETOL-armen (N=1019) og 1 i PegIntron 1 mcg/REBETOL-armen (N= 1016), og 6 av disse forekom hos personer som fikk Pegasys/Copegus (N=1035); det var 3 selvmord som skjedde under oppfølgingsperioden utenfor behandlingen hos personer som fikk PegIntron (1,5 mcg/kg)/REBETOL 200 mg kombinasjonsbehandling.
studie 1 og 2 avbrøt 10 til 14 % av pasientene som fikk PegIntron, alene eller i kombinasjon med REBETOL 200 mg, behandlingen sammenlignet med 6 % behandlet med INTRON A alene og 13 % behandlet med INTRON A i kombinasjon med REBETOL. Tilsvarende i studie 3 avbrøt 15 % av pasientene som fikk PegIntron i kombinasjon med vektbasert REBETOL og 14 % av pasientene som fikk PegIntron og flatdose REBETOL 200 mg behandlingen på grunn av en bivirkning. De vanligste årsakene til seponering av behandlingen var relatert til kjente interferoneffekter av psykiatriske, systemiske (f.eks. tretthet, hodepine) eller gastrointestinale bivirkninger. I studie 4 ble 13 % av forsøkspersonene i PegIntron 1,5 mcg/REBETOL 200 mg-armen, 10 % i PegIntron 1 mcg/REBETOL 200 mg-armen og 13 % i Pegasys 180 mcg/Copegus-armen avbrutt på grunn av uønskede hendelser.
studie 2 forekom dosereduksjoner på grunn av bivirkninger hos 42 % av pasientene som fikk PegIntron (1,5 mcg/kg)/REBETOL 200 mg og hos 34 % av de som fikk INTRON A/REBETOL. De fleste forsøkspersoner (57 %) som veide 60 kg eller mindre som fikk PegIntron (1,5 mcg/kg)/REBETOL trengte dosereduksjon. Reduksjon av interferon var doserelatert (PegIntron 1,5 mcg/kg større enn PegIntron 0,5 mcg/kg eller INTRON A), henholdsvis 40 %, 27 %, 28 %. Dosereduksjonen for REBETOL 200 mg var lik for alle tre gruppene, 33 til 35 %. De vanligste årsakene til doseendringer var nøytropeni (18 %) eller anemi (9 %) (se Laboratorieverdier ). Andre vanlige årsaker inkluderer depresjon, tretthet, kvalme og trombocytopeni. I studie 3 forekom doseendringer på grunn av bivirkninger hyppigere med vektbasert dosering (WBD) sammenlignet med flat dosering (henholdsvis 29 % og 23 %). I studie 4 hadde 16 % av forsøkspersonene en dosereduksjon av PegIntron til 1 mcg/kg i kombinasjon med REBETOL 200 mg, med ytterligere 4 % som krevde den andre dosereduksjonen av PegIntron til 0,5 mcg/kg på grunn av bivirkninger sammenlignet med 15 % av forsøkspersoner i Pegasys/Copegus-armen, som trengte en dosereduksjon til 135 mcg/uke med Pegasys, med ytterligere 7 % i Pegasys/Copegus-armen som krevde andre dosereduksjon til 90 mcg/uke med Pegasys.
kombinasjonsstudiene PegIntron/REBETOL 200 mg var de vanligste bivirkningene psykiatriske, som forekom blant 77 % av forsøkspersonene i studie 2 og 68 % til 69 % av forsøkspersonene i studie 3. Disse psykiatriske bivirkningene inkluderte oftest depresjon, irritabilitet og søvnløshet, hver rapportert av omtrent 30 % til 40 % av pasientene i alle behandlingsgrupper. Selvmordsatferd (ideer, forsøk og selvmord) forekom hos 2 % av alle forsøkspersoner under behandling eller under oppfølging etter avsluttet behandling [se ADVARSLER OG FORHOLDSREGLER ]. I studie 4 forekom psykiatriske bivirkninger hos 58 % av forsøkspersonene i PegIntron 1,5 mcg/REBETOL 200mg-armen, 55 % av forsøkspersonene i PegIntron 1 mcg/REBETOL 200mg-armen og 57 % av forsøkspersonene i Pegasys-180mcg-armen. .
PegIntron induserte tretthet eller hodepine hos omtrent to tredjedeler av forsøkspersonene, med feber eller stivhet hos omtrent halvparten av forsøkspersonene. Alvorlighetsgraden av noen av disse systemiske symptomene (f.eks. feber og hodepine) hadde en tendens til å avta etter hvert som behandlingen fortsatte. I studie 1 og 2 oppsto betennelse og reaksjon på påføringsstedet (f.eks. blåmerker, kløe og irritasjon) med omtrent dobbelt så høy forekomst med PegIntron-behandlinger (hos opptil 75 % av forsøkspersonene) sammenlignet med INTRON A. Smerte på injeksjonsstedet var imidlertid sjelden (2 til 3 %) i alle grupper. I studie 3 var det en total forekomst på 23 % til 24 % for reaksjoner på injeksjonsstedet eller betennelse.
Pasienter som fikk REBETOL/PegIntron som re-behandling etter å ha mislyktes i et tidligere interferonkombinasjonsregime, rapporterte bivirkninger tilsvarende de som tidligere var assosiert med dette regimet under kliniske studier med behandlingsnaive forsøkspersoner.
Pediatriske fag
Generelt var bivirkningsprofilen i den pediatriske populasjonen lik den som ble observert hos voksne. I den pediatriske studien var de mest utbredte bivirkningene hos alle forsøkspersoner pyreksi (80 %), hodepine (62 %), nøytropeni (33 %), tretthet (30 %), anoreksi (29 %), erytem på injeksjonsstedet (29 %) og oppkast (27 %). De fleste bivirkningene rapportert i studien var milde eller moderate i alvorlighetsgrad. Alvorlige bivirkninger ble rapportert hos 7 % (8/107) av alle forsøkspersoner og inkluderte smerter på injeksjonsstedet (1 %), smerter i ekstremiteter (1 %), hodepine (1 %), nøytropeni (1 %) og pyreksi (4 %). Viktige bivirkninger som oppsto i denne forsøkspopulasjonen var nervøsitet (7 %; 7/107), aggresjon (3 %; 3/107), sinne (2 %; 2/107) og depresjon (1 %; 1/107) . Fem forsøkspersoner fikk levotyroksinbehandling, tre med klinisk hypotyreose og to med asymptomatiske TSH-økninger. Vekt- og høydeøkning hos pediatriske forsøkspersoner behandlet med PegIntron pluss REBETOL lå bak det som ble forutsagt av normative populasjonsdata for hele behandlingslengden. Alvorlig hemmet veksthastighet (mindre enn 3. persentil) ble observert hos 70 % av pasientene under behandling.
Doseendringer av PegIntron og/eller ribavirin var nødvendig hos 25 % av pasientene på grunn av behandlingsrelaterte bivirkninger, oftest for anemi, nøytropeni og vekttap. To forsøkspersoner (2 %; 2/107) avbrøt behandlingen som følge av en bivirkning.
Bivirkninger som oppsto med en forekomst på over eller lik 10 % hos de pediatriske forsøkspersonene er gitt i tabell 7.
94 av 107 forsøkspersoner ble registrert i en 5-årig langsiktig oppfølgingsstudie. Langtidseffekten på vekst var mindre hos de som ble behandlet i 24 uker enn de som ble behandlet i 48 uker. Tjuefire prosent av forsøkspersonene (11/46) behandlet i 24 uker og 40 % av forsøkspersonene (19/48) behandlet i 48 uker hadde en reduksjon på > 15 persentil høyde for alder fra førbehandling til slutten av 5 år langsiktig oppfølging sammenlignet med før behandlings baseline persentiler. Elleve prosent av forsøkspersonene (5/46) behandlet i 24 uker og 13 % av forsøkspersonene (6/48) behandlet i 48 uker ble observert å ha en nedgang fra før-behandlingens baseline på > 30 høyde-for-alder persentiler til slutten av 5 års langtidsoppfølging. Mens det ble observert på tvers av alle aldersgrupper, så den høyeste risikoen for redusert høyde ved slutten av langtidsoppfølgingen ut til å korrelere med oppstart av kombinasjonsbehandling i løpet av årene med forventet toppveksthastighet. [Se ADVARSLER OG FORHOLDSREGLER ]
Laboratorieverdier
Voksne og pediatriske fag
Bivirkningsprofilen i studie 3, som sammenlignet PegIntron/vektbasert REBETOL 200 mg-kombinasjon med en PegIntron/flat dose REBETOL 200mg-regime, avslørte en økt forekomst av anemi med vektbasert dosering (29 % vs. 19 % for vektbaserte doser). vs. flatdoseregimer, henholdsvis). Imidlertid var flertallet av tilfellene av anemi milde og responderte på dosereduksjoner.
Endringer i utvalgte laboratorieverdier under behandling i kombinasjon med REBETOL 200 mg behandling er beskrevet nedenfor. Nedgang i hemoglobin, leukocytter, nøytrofiler og blodplater kan kreve dosereduksjon eller permanent seponering av behandlingen [se DOSERING OG ADMINISTRASJON ]. Endringer i utvalgte laboratorieverdier under behandlingen er beskrevet i tabell 8. De fleste endringene i laboratorieverdier i PegIntron/REBETOL 200 mg studien med pediatri var milde eller moderate.
Hemoglobin
Hemoglobinnivået sank til mindre enn 11 g/dL hos omtrent 30 % av forsøkspersonene i studie 2. I studie 3 hadde 47 % av forsøkspersonene som fikk WBD REBETOL 200mg og 33 % på flatdose REBETOL 200mg reduksjoner i hemoglobinnivåer på mindre enn 11 g /dl. Reduksjoner i hemoglobin til mindre enn 9 g/dL forekom hyppigere hos personer som fikk WBD sammenlignet med flat dosering (henholdsvis 4 % og 2 %). I studie 2 var doseendring nødvendig hos 9 % og 13 % av pasientene i gruppene PegIntron/REBETOL 200 mg og INTRON A/REBETOL 200 mg. I studie 4 hadde forsøkspersoner som fikk PegIntron (1,5 mcg/kg)/REBETOL reduksjoner i hemoglobinnivåer til mellom 8,5 til mindre enn 10 g/dL (28 %) og til mindre enn 8,5 g/dL (3 %), mens hos pasienter Ved å få Pegasys 180 mcg/Copegus skjedde disse reduksjonene hos henholdsvis 26 % og 4 % av pasientene. Hemoglobinnivået ble stabilt ved behandlingsuke 4-6 i gjennomsnitt. Det typiske mønsteret som ble observert var en reduksjon i hemoglobinnivået ved behandling uke 4 etterfulgt av stabilisering og et platå, som ble opprettholdt til slutten av behandlingen. I PegIntron monoterapistudien var hemoglobinreduksjoner generelt milde og doseendringer var sjelden nødvendig [se DOSERING OG ADMINISTRASJON ].
Nøytrofiler
Nedgang i nøytrofiltall ble observert hos et flertall av voksne pasienter behandlet med kombinasjonsbehandling med REBETOL 200 mg i studie 2 (85 %) og INTRON A/REBETOL (60 %). Alvorlig potensielt livstruende nøytropeni (mindre enn 0,5 x 109/L) forekom hos 2 % av pasientene behandlet med INTRON A/REBETOL 200 mg og hos ca. 4 % av pasientene behandlet med PegIntron/REBETOL 200 mg i studie 2. 18 prosent av pasientene. PegIntron/REBETOL i studie 2 krevde modifikasjon av interferondosering. Få forsøkspersoner (mindre enn 1 %) krevde permanent seponering av behandlingen. Nøytrofiltall returnerte vanligvis til nivåer før behandling 4 uker etter avsluttet behandling [se DOSERING OG ADMINISTRASJON ].
Blodplater
Blodplateantallet sank til mindre enn 100 000/mm³ hos ca. 20 % av pasientene behandlet med PegIntron alene eller med REBETOL 200 mg og hos 6 % av voksne pasienter behandlet med INTRON A/REBETOL. Alvorlig reduksjon i antall blodplater (mindre enn 50 000/mm³) forekommer hos mindre enn 4 % av voksne personer. Pasienter kan trenge seponering eller doseendring som følge av blodplatereduksjon [se DOSERING OG ADMINISTRASJON ]. I studie 2 krevde 1 % eller 3 % av forsøkspersonene doseendring av henholdsvis INTRON A eller PegIntron. Blodplateantallet returnerte vanligvis til nivåer før behandling 4 uker etter avsluttet behandling.
Skjoldbrusk funksjon
Utvikling av TSH-avvik, med eller uten kliniske manifestasjoner, er assosiert med interferonbehandlinger. I studie 2 forekom klinisk tilsynelatende skjoldbruskkjertelforstyrrelser blant forsøkspersoner behandlet med enten INTRON A eller PegIntron (med eller uten REBETOL) med lignende forekomst (5 % for hypotyreose og 3 % for hypertyreose). Forsøkspersonene utviklet nye TSH-avvik under behandling og i oppfølgingsperioden. Ved slutten av oppfølgingsperioden hadde 7 % av forsøkspersonene fortsatt unormale TSH-verdier.
Bilirubin og urinsyre
I studie 2 utviklet 10 til 14 % av forsøkspersonene hyperbilirubinemi og 33 til 38 % utviklet hyperurikemi i forbindelse med hemolyse. Seks forsøkspersoner utviklet mild til moderat gikt.
Erfaring fra kliniske studier – REBETOL/INTRON En kombinasjonsterapi
Voksne emner
kliniske studier avbrøt henholdsvis 19 % og 6 % av tidligere ubehandlede og tilbakefallspasienter behandlingen på grunn av bivirkninger i kombinasjonsarmene sammenlignet med 13 % og 3 % i interferonarmene. Utvalgte behandlingsrelaterte bivirkninger som oppstod i USA-studiene med større enn eller lik 5 % forekomst er gitt etter behandlingsgruppe (se tabell 9). Generelt ble de utvalgte behandlingsrelaterte bivirkningene rapportert med lavere forekomst i de internasjonale studiene sammenlignet med de amerikanske studiene, med unntak av asteni, influensalignende symptomer, nervøsitet og kløe.
Pediatriske fag
kliniske studier med 118 pediatriske personer i alderen 3 til 16 år, avbrøt 6 % behandlingen på grunn av bivirkninger. Doseendringer var nødvendig hos 30 % av pasientene, oftest for anemi og nøytropeni. Generelt var bivirkningsprofilen i den pediatriske populasjonen lik den som ble observert hos voksne. Forstyrrelser på injeksjonsstedet, feber, anoreksi, oppkast og emosjonell labilitet forekom hyppigere hos pediatriske forsøkspersoner sammenlignet med voksne. Omvendt opplevde pediatriske personer mindre tretthet, dyspepsi, artralgi, søvnløshet, irritabilitet, nedsatt konsentrasjon, dyspné og pruritus sammenlignet med voksne. Utvalgte behandlingsrelaterte bivirkninger som oppstod med større enn eller lik 5 % forekomst blant alle pediatriske forsøkspersoner som fikk den anbefalte dosen av REBETOL/INTRON A kombinasjonsbehandling er gitt i tabell 9.
løpet av et 48-ukers behandlingsforløp var det en reduksjon i hastigheten av lineær vekst (gjennomsnittlig prosentiltilordning reduksjon på 7 %) og en reduksjon i frekvensen av vektøkning (gjennomsnittlig prosentiltilordning reduksjon på 9 %). En generell reversering av disse trendene ble notert i løpet av den 24 uker lange perioden etter behandling. Langtidsdata fra et begrenset antall pasienter antyder imidlertid at kombinasjonsbehandling kan indusere en vekstinhibering som resulterer i redusert endelig voksenhøyde hos noen pasienter [se ADVARSLER OG FORHOLDSREGLER ].
Laboratorieverdier
Endringer i utvalgte hematologiske verdier (hemoglobin, hvite blodlegemer, nøytrofiler og blodplater) under behandlingen er beskrevet nedenfor (se tabell 10).
Hemoglobin. Hemoglobinnedgang blant forsøkspersoner som fikk REBETOL-behandling begynte ved uke 1, med stabilisering innen uke 4. Hos tidligere ubehandlede forsøkspersoner behandlet i 48 uker var gjennomsnittlig maksimal reduksjon fra baseline 3,1 g/dL i den amerikanske studien og 2,9 g/dL i den internasjonale prøve. Hos personer med tilbakefall var gjennomsnittlig maksimal reduksjon fra baseline 2,8 g/dL i den amerikanske studien og 2,6 g/dL i den internasjonale studien. Hemoglobinverdiene gikk tilbake til nivåer før behandling innen 4 til 8 uker etter avsluttet behandling hos de fleste pasientene.
Bilirubin og urinsyre. Økning i både bilirubin og urinsyre, assosiert med hemolyse, ble observert i kliniske studier. De fleste var moderate biokjemiske endringer og ble reversert innen 4 uker etter avsluttet behandling. Denne observasjonen skjedde oftest hos personer med en tidligere diagnose av Gilberts syndrom. Dette har ikke vært assosiert med leverdysfunksjon eller klinisk morbiditet.
Postmarketing-opplevelser
Følgende bivirkninger er identifisert og rapportert under bruk av REBETOL i kombinasjon med INTRON A eller PegIntron etter godkjenning. Fordi disse reaksjonene rapporteres frivillig fra en populasjon av usikker størrelse, er det ikke alltid mulig å pålitelig estimere frekvensen deres eller etablere en årsakssammenheng med legemiddeleksponering.
Blod- og lymfesystemlidelser
Ren rødcelleaplasi, aplastisk anemi
Øre- og labyrintlidelser
Hørselsforstyrrelser, svimmelhet
Respiratoriske, thorax- og mediastinumsykdommer
Pulmonal hypertensjon
Øyelidelser
Serøs netthinneløsning
Endokrine lidelser
Diabetes
NARKOTIKAHANDEL
Didanosin
Eksponering for didanosin eller dets aktive metabolitt (dideoksyadenosin 5'-trifosfat) øker når didanosin gis samtidig med ribavirin, noe som kan forårsake eller forverre klinisk toksisitet; Derfor er samtidig administrering av REBETOL 200 mg kapsler eller mikstur og didanosin kontraindisert. Rapporter om fatal leversvikt, samt perifer nevropati, pankreatitt og symptomatisk hyperlaktatemi/laktacidose er rapportert i kliniske studier.
Nukleosid-analoger
Leverdekompensasjon (noen dødelig) har forekommet hos cirrhotiske HIV/HCV-infiserte pasienter som får antiretroviral kombinasjonsbehandling for HIV og interferon alfa og ribavirin. Tilsetning av behandling med alfa-interferoner alene eller i kombinasjon med ribavirin kan øke risikoen i denne pasientpopulasjonen. Pasienter som får interferon med ribavirin og nukleosid revers transkriptasehemmere (NRTIer) bør overvåkes nøye for behandlingsassosierte toksisiteter, spesielt leverdekompensasjon og anemi. Seponering av NRTIer bør anses som medisinsk hensiktsmessig (se merking for individuelle NRTI-produkter ). Dosereduksjon eller seponering av interferon, ribavirin eller begge bør også vurderes hvis forverrede kliniske toksisiteter observeres, inkludert leverdekompensasjon (f.eks. Child-Pugh større enn 6).
Ribavirin kan antagonisere cellekulturens antivirale aktivitet til stavudin og zidovudin mot HIV. Ribavirin har vist seg i cellekultur å hemme fosforylering av lamivudin, stavudin og zidovudin, noe som kan føre til redusert antiretroviral aktivitet. I en studie med et annet pegylert interferon i kombinasjon med ribavirin ble det imidlertid ikke observert noen farmakokinetisk (f.eks. plasmakonsentrasjoner eller intracellulære trifosforylerte aktive metabolittkonsentrasjoner) eller farmakodynamiske (f.eks. tap av HIV/HCV virologisk undertrykkelse) interaksjon når ribavirin og lamivudin (n) =18), stavudin (n=10) eller zidovudin (n=6) ble administrert samtidig som en del av et multilegemiddelregime hos HIV/HCV-infiserte personer. Derfor bør samtidig bruk av ribavirin med ett av disse legemidlene brukes med forsiktighet.
Legemidler metabolisert av Cytokrom P-450
Resultater av in vitro-studier med både humane og rottelevermikrosompreparater indikerte liten eller ingen cytokrom P-450 enzymmediert metabolisme av ribavirin, med minimalt potensial for P-450 enzymbaserte legemiddelinteraksjoner.
Det ble ikke observert noen farmakokinetiske interaksjoner mellom INTRON A og REBETOL 200 mg kapsler i en farmakokinetisk flerdosestudie.
Azatioprin
Bruk av ribavirin til behandling av kronisk hepatitt C hos pasienter som får azatioprin har blitt rapportert å indusere alvorlig pancytopeni og kan øke risikoen for azatioprin-relatert myelotoksisitet. Inosinmonofosfatdehydrogenase (IMDH) er nødvendig for en av metabolske veier til azatioprin. Ribavirin er kjent for å hemme IMDH, og dermed føre til akkumulering av en azatioprinmetabolitt, 6-metyltioinosinmonofosfat (6-MTITP), som er assosiert med myelotoksisitet (nøytropeni, trombocytopeni og anemi). Pasienter som får azatioprin sammen med ribavirin bør ha fullstendige blodtellinger, inkludert blodplatetall, overvåket ukentlig den første måneden, to ganger månedlig i den andre og tredje måneden av behandlingen, deretter månedlig eller oftere hvis dosendringer eller andre behandlingsendringer er nødvendig [se ADVARSLER OG FORHOLDSREGLER ].
ADVARSLER
Inkludert som en del av FORHOLDSREGLER seksjon.
FORHOLDSREGLER
Svangerskap
REBETOL 200mg kapsler og mikstur kan forårsake fødselsskader og død hos det ufødte barnet. Behandling med REBETOL bør ikke startes før en rapport om en negativ graviditetstest er innhentet rett før planlagt behandlingsstart. Pasienter bør bruke minst to former for prevensjon og ha månedlige graviditetstester under behandlingen og i 6-månedersperioden etter at behandlingen er avsluttet. Ekstrem forsiktighet må utvises for å unngå graviditet hos kvinnelige pasienter og hos kvinnelige partnere til mannlige pasienter. REBETOL 200mg har demonstrert signifikante teratogene og embryocidale effekter hos alle dyrearter der tilstrekkelige studier er utført. Disse effektene oppstod ved doser som lavt som en tjuendedel av den anbefalte humane dosen av ribavirin. Behandling med REBETOL 200 mg bør ikke startes før en rapport om en negativ graviditetstest er innhentet umiddelbart før planlagt oppstart av terapi [se ADVARSEL i boks , KONTRAINDIKASJONER , Bruk i spesifikke populasjoner , og PASIENTINFORMASJON ].
Anemi
Den primære toksisiteten til ribavirin er hemolytisk anemi, som ble observert hos ca. 10 % av REBETOL/INTRON A-behandlede personer i kliniske studier. Anemien assosiert med REBETOL kapsler oppstår innen 1 til 2 uker etter oppstart av behandlingen. Fordi det første fallet i hemoglobin kan være betydelig, anbefales det at hemoglobin eller hematokrit oppnås før behandlingsstart og ved uke 2 og uke 4 av behandlingen, eller oftere hvis det er klinisk indisert. Pasienter bør deretter følges som klinisk hensiktsmessig [se DOSERING OG ADMINISTRASJON ].
Fatale og ikke-dødelige hjerteinfarkter er rapportert hos pasienter med anemi forårsaket av REBETOL. Pasienter bør vurderes for underliggende hjertesykdom før oppstart av ribavirinbehandling. Pasienter med allerede eksisterende hjertesykdom bør ha elektrokardiogram administrert før behandling, og bør overvåkes på passende måte under behandlingen. Hvis det er noen forverring av kardiovaskulær status, bør behandlingen avbrytes eller avbrytes [se DOSERING OG ADMINISTRASJON ]. Fordi hjertesykdom kan forverres av legemiddelindusert anemi, bør pasienter med en historie med betydelig eller ustabil hjertesykdom ikke bruke REBETOL.
Pankreatitt
Behandling med REBETOL og INTRON A eller PegIntron bør avbrytes hos pasienter med tegn og symptomer på pankreatitt og seponeres hos pasienter med bekreftet pankreatitt.
Lungelidelser
Lungesymptomer, inkludert dyspné, lungeinfiltrater, pneumonitt, pulmonal hypertensjon og lungebetennelse, er rapportert under behandling med REBETOL med alfa-interferon kombinasjonsbehandling; sporadiske tilfeller av dødelig lungebetennelse har forekommet. I tillegg er det rapportert om sarkoidose eller forverring av sarkoidose. Hvis det er tegn på lungeinfiltrater eller lungefunksjonssvikt, bør pasienten overvåkes nøye, og kombinasjonsbehandlingen bør seponeres om nødvendig.
Oftalmologiske lidelser
Ribavirin brukes i kombinasjonsbehandling med alfa-interferoner. Nedsatt eller tap av syn, retinopati inkludert makulaødem, retinal arterie eller vene, trombose, netthinneblødninger og bomullsdottflekker, optikusnevritt, papilleødem og serøs netthinneløsning induseres eller forverres ved behandling med alfa-interferoner. Alle pasienter bør få en øyeundersøkelse ved baseline. Pasienter med allerede eksisterende oftalmologiske lidelser (f.eks. diabetisk eller hypertensiv retinopati) bør få periodiske oftalmologiske undersøkelser under kombinasjonsbehandling med alfa-interferonbehandling. Enhver pasient som utvikler okulære symptomer bør få en rask og fullstendig øyeundersøkelse. Kombinasjonsbehandling med alfa-interferoner bør seponeres hos pasienter som utvikler nye eller forverrede oftalmologiske lidelser.
Laboratorietester
PegIntron i kombinasjon med ribavirin kan forårsake alvorlig reduksjon i antall nøytrofiler og blodplater, og hematologiske, endokrine (f.eks. TSH) og hepatiske abnormiteter.
Pasienter på PegIntron/REBETOL kombinasjonsbehandling bør ha hematologi og blodkjemitesting før behandlingsstart og deretter med jevne mellomrom. I den kliniske studien for voksne ble fullstendig blodtelling (inkludert hemoglobin-, nøytrofil- og blodplatetall) og kjemi (inkludert ASAT, ALT, bilirubin og urinsyre) målt i løpet av behandlingsperioden i uke 2, 4, 8, 12 og deretter med 6-ukers intervaller, eller oftere hvis det utviklet seg abnormiteter. Hos pediatriske forsøkspersoner ble de samme laboratorieparametrene evaluert med tilleggsvurdering av hemoglobin ved behandlingsuke 6. TSH-nivåer ble målt hver 12. uke i behandlingsperioden. HCV-RNA bør måles med jevne mellomrom under behandlingen [se DOSERING OG ADMINISTRASJON ].
Tann- og periodontale lidelser
Tann- og periodontale lidelser er rapportert hos pasienter som får kombinasjonsbehandling med ribavirin og interferon eller peginterferon. I tillegg kan munntørrhet ha en skadelig effekt på tenner og slimhinner i munnen under langtidsbehandling med kombinasjonen REBETOL og pegylert eller ikke-pegylert interferon alfa-2b. Pasienter bør pusse tennene grundig to ganger daglig og ha regelmessige tannundersøkelser. Hvis det oppstår brekninger, bør de rådes til å skylle munnen grundig etterpå.
Samtidig administrering av Azatioprin
Pancytopeni (markert reduksjon i røde blodlegemer, nøytrofiler og blodplater) og benmargssuppresjon er rapportert i litteraturen å oppstå innen 3 til 7 uker etter samtidig administrering av pegylert interferon/ribavirin og azatioprin. Hos dette begrensede antallet pasienter (n=8) var myelotoksisiteten reversibel innen 4 til 6 uker etter seponering av både HCV antiviral behandling og samtidig azatioprin og kom ikke tilbake ved gjeninnføring av noen av behandlingene alene. PegIntron, REBETOL og azatioprin bør seponeres for pancytopeni, og pegylert interferon/ribavirin bør ikke gjeninnføres med samtidig azatioprin [se NARKOTIKAHANDEL ].
Påvirkning på vekst – pediatrisk bruk
Data om effekten av PegIntron og REBETOL på vekst kommer fra en åpen studie på forsøkspersoner i alderen 3 til 17 år, der vekt- og høydeendringer sammenlignes med amerikanske normative befolkningsdata. Generelt henger vekt- og høydeøkningen til pediatriske forsøkspersoner behandlet med PegIntron og REBETOL 200 mg bak det som er forutsagt av normative populasjonsdata for hele behandlingslengden. Alvorlig hemmet veksthastighet (mindre enn 3. persentil) ble observert hos 70 % av pasientene under behandling. Etter behandling forekom rebound-vekst og vektøkning hos de fleste pasientene. Langtidsoppfølgingsdata hos pediatriske personer indikerer imidlertid at PegIntron i kombinasjonsbehandling med REBETOL kan indusere en vekstinhibering som resulterer i redusert voksenhøyde hos noen pasienter [se BIVIRKNINGER ].
På samme måte ble en påvirkning på vekst sett hos forsøkspersoner etter behandling med REBETOL 200 mg og INTRON A kombinasjonsbehandling i ett år. I en langsiktig oppfølgingsstudie av et begrenset antall av disse forsøkspersonene, resulterte kombinasjonsbehandling i redusert endelig voksenhøyde hos noen forsøkspersoner [se BIVIRKNINGER ].
Bruksbeskyttelse
Basert på resultater fra kliniske studier, er ribavirin monoterapi ikke effektiv for behandling av kronisk hepatitt C-virusinfeksjon; derfor må ikke REBETOL 200 mg kapsler eller mikstur brukes alene. Sikkerheten og effekten av REBETOL kapsler og mikstur er kun fastslått når de brukes sammen med INTRON A eller PegIntron (ikke andre interferoner) som kombinasjonsbehandling.
Sikkerhet og effekt av behandling med REBETOL/INTRON A og PegIntron for behandling av HIV-infeksjon, adenovirus, RSV, parainfluensa eller influensainfeksjoner er ikke fastslått. REBETOL 200mg kapsler skal ikke brukes til disse indikasjonene. Ribavirin for inhalasjon har egen merking, som bør konsulteres dersom ribavirin inhalasjonsbehandling vurderes.
Det er betydelige bivirkninger forårsaket av behandling med REBETOL/INTRON A eller PegIntron, inkludert alvorlig depresjon og selvmordstanker, hemolytisk anemi, undertrykkelse av benmargsfunksjon, autoimmune og infeksjonssykdommer, lungedysfunksjon, pankreatitt og diabetes. Selvmordstanker eller selvmordsforsøk forekom hyppigere blant pediatriske pasienter, primært ungdom, sammenlignet med voksne pasienter (2,4 % versus 1 %) under behandling og oppfølging utenfor behandlingen. Merking av INTRON A og PegIntron bør gjennomgås i sin helhet for ytterligere sikkerhetsinformasjon før oppstart av kombinasjonsbehandling.
Informasjon om pasientveiledning
Råd pasienten til å lese FDA-godkjent pasientmerking ( Medisinveiledning ).
Anemi
Den vanligste bivirkningen som oppstår med REBETOL kapsler er anemi, som kan være alvorlig [se ADVARSLER OG FORHOLDSREGLER og BIVIRKNINGER ]. Pasienter bør informeres om at laboratorieevalueringer er nødvendig før behandlingsstart og med jevne mellomrom etterpå [se DOSERING OG ADMINISTRASJON ]. Det anbefales at pasienter er godt hydrert, spesielt i de innledende stadiene av behandlingen.
Svangerskap
Pasienter må informeres om at REBETOL 200 mg kapsler og mikstur kan forårsake fødselsskader og død hos det ufødte barnet. REBETOL 200mg må ikke brukes av kvinner som er gravide eller av menn hvis kvinnelige partnere er gravide. Ekstrem forsiktighet må utvises for å unngå graviditet hos kvinnelige pasienter og hos kvinnelige partnere til mannlige pasienter som tar REBETOL. REBETOL skal ikke startes før en rapport om en negativ graviditetstest er innhentet rett før behandlingsstart. Pasienter må utføre en graviditetstest månedlig under behandlingen og i 6 måneder etter behandlingen.
Kvinner i fertil alder må veiledes om bruk av effektiv prevensjon (to pålitelige former) før behandlingsstart. Pasienter (menn og kvinner) må informeres om teratogene/embryocidale risikoer og må instrueres om å bruke effektiv prevensjon under REBETOL 200 mg og i 6 måneder etter behandling. Pasienter (mannlige og kvinnelige) bør rådes til å varsle legen umiddelbart i tilfelle graviditet [se KONTRAINDIKASJONER , ADVARSLER OG FORHOLDSREGLER , og Bruk i spesifikke populasjoner ].
Hvis graviditet oppstår under behandling eller i løpet av 6 måneder etter behandling, må pasienten informeres om den teratogene risikoen ved behandling med REBETOL 200 mg for fosteret. Pasienter, eller partnere til pasienter, bør umiddelbart rapportere enhver graviditet som oppstår under behandling eller innen 6 måneder etter avsluttet behandling til legen. Forskrivere bør rapportere slike tilfeller ved å ringe 1-800-593-2214.
Risikoer versus fordeler
Pasienter som får REBETOL kapsler bør informeres om fordelene og risikoene forbundet med behandlingen, instrueres i riktig bruk og henvises til pasienten Medisinveiledning . Pasienter bør informeres om at effekten av behandling av hepatitt C-infeksjon på overføring ikke er kjent, og at passende forholdsregler for å forhindre overføring av hepatitt C-virus bør tas.
Pasienter bør informeres om hva de skal gjøre dersom de glemmer en dose REBETOL; den glemte dosen bør tas så snart som mulig i løpet av samme dag. Pasienter bør ikke doble neste dose. Pasienter bør rådes til å kontakte helsepersonell dersom de har spørsmål.
Ikke-klinisk toksikologi
Karsinogenese, mutagenese, svekkelse av fruktbarhet
Karsinogenese
Ribavirin forårsaket ikke en økning i noen tumortype når det ble administrert i 6 måneder i den transgene p53-mangelfulle musemodellen ved doser opp til 300 mg/kg (estimert humanekvivalent på 25 mg/kg basert på justering av kroppsoverflate for en voksen på 60 kg 1,9 ganger den maksimale anbefalte daglige dosen til mennesker). Ribavirin var ikke-karsinogent når det ble administrert i 2 år til rotter i doser opptil 40 mg/kg (estimert humanekvivalent på 5,71 mg/kg basert på kroppsoverflatejustering for en voksen på 60 kg).
Mutagenese
Ribavirin viste økt forekomst av mutasjoner og celletransformasjon i flere genotoksisitetsanalyser. Ribavirin var aktiv i Balb/3T3 in vitro celletransformasjonsanalyse. Mutagen aktivitet ble observert i muselymfomanalysen, og ved doser på 20 til 200 mg/kg (estimert humanekvivalent på 1,67 til 16,7 mg/kg, basert på kroppsoverflatejustering for en voksen på 60 kg; 0,1 til 1 ganger maksimum anbefalt human 24-timers dose ribavirin) i en mikronukleusanalyse hos mus. En dominant dødelig analyse hos rotter var negativ, noe som indikerer at hvis mutasjoner oppsto i rotter, ble de ikke overført gjennom hannkjønnsceller.
Nedsatt fertilitet
Ribavirin viste signifikante embryocidale og teratogene effekter ved doser godt under anbefalt human dose hos alle dyrearter der tilstrekkelige studier er utført. Misdannelser i hodeskallen, ganen, øyet, kjeven, lemmer, skjelett og mage-tarmkanalen ble notert. Forekomsten og alvorlighetsgraden av teratogene effekter økte med eskalering av medikamentdosen. Overlevelsen av fostre og avkom ble redusert. I konvensjonelle embryotoksisitets-/teratogenesitetsstudier på rotter og kaniner, var de observerte dosenivåene uten effekt godt under de for foreslått klinisk bruk (0,3 mg/kg/dag for både rotte og kanin; omtrent 0,06 ganger den anbefalte 24-timersdosen til mennesker av ribavirin). Ingen maternell toksisitet eller effekter på avkom ble observert i en peri/postnatal toksisitetsstudie på rotter doseret oralt med opptil 1 mg/kg/dag (estimert human ekvivalent dose på 0,17 mg/kg basert på kroppsoverflatejustering for en voksen på 60 kg ; ca. 0,01 ganger den maksimale anbefalte humane 24-timers dosen av ribavirin) [se KONTRAINDIKASJONER , og ADVARSLER OG FORHOLDSREGLER ].
Fertile kvinner og partnere til fertile kvinner bør ikke få REBETOL med mindre pasienten og hans/hennes partner bruker effektiv prevensjon (to pålitelige former). Basert på en flerdosehalveringstid (t1/2) av ribavirin på 12 dager, må effektiv prevensjon brukes i 6 måneder etter behandling (f.eks. 15 halveringstider for clearance for ribavirin).
REBETOL bør brukes med forsiktighet hos fertile menn. I studier på mus for å evaluere tidsforløpet og reversibiliteten av ribavirin-indusert testikkeldegenerasjon ved doser på 15 til 150 mg/kg/dag (estimert humanekvivalent på 1,25 til 12,5 mg/kg/dag, basert på kroppsoverflatejustering for en 60 kg voksen; 0,1-0,8 ganger den maksimale humane 24-timers dosen av ribavirin) administrert i 3 eller 6 måneder, oppstod abnormiteter i sædceller. Ved seponering av behandlingen var i det vesentlige total bedring etter ribavirinindusert testikkeltoksisitet tydelig innen 1 eller 2 spermatogenesesykluser.
Bruk i spesifikke populasjoner
Svangerskap
Graviditetskategori X
[Se KONTRAINDIKASJONER , ADVARSLER OG FORHOLDSREGLER , og Ikke-klinisk toksikologi ].
Behandling og etterbehandling:
Potensiell risiko for fosteret
Ribavirin er kjent for å akkumulere i intracellulære komponenter hvorfra det fjernes veldig sakte. Det er ikke kjent om ribavirin i sædceller vil ha en potensiell teratogene effekt ved befruktning av eggene. I en studie på rotter ble det konkludert med at dominant dødelighet ikke ble indusert av ribavirin ved doser opp til 200 mg/kg i 5 dager (estimerte humanekvivalente doser på 7,14 til 28,6 mg/kg, basert på kroppsoverflatejustering for 60 kg voksen; opptil 1,7 ganger den maksimale anbefalte humane dosen av ribavirin). Men på grunn av de potensielle humane teratogene effektene av ribavirin, bør mannlige pasienter rådes til å ta alle forholdsregler for å unngå risiko for graviditet for sine kvinnelige partnere.
Kvinner i fertil alder bør ikke få REBETOL med mindre de bruker effektiv prevensjon (to pålitelige former) i løpet av behandlingsperioden. I tillegg bør effektiv prevensjon brukes i 6 måneder etter behandling basert på en flerdosehalveringstid (t1/2) av ribavirin på 12 dager.
Mannlige pasienter og deres kvinnelige partnere må praktisere effektiv prevensjon (to pålitelige former) under behandling med REBETOL og i 6 måneder etter terapiperioden (f.eks. 15 halveringstider for ribavirin-clearance fra kroppen).
Et Ribavirin-graviditetsregister er etablert for å overvåke mors-føtale utfall av graviditeter hos kvinnelige pasienter og kvinnelige partnere til mannlige pasienter eksponert for ribavirin under behandling og i 6 måneder etter avsluttet behandling. Leger og pasienter oppfordres til å rapportere slike tilfeller ved å ringe 1-800-593-2214.
Ammende mødre
Det er ikke kjent om REBETOL-produktet skilles ut i morsmelk. På grunn av potensialet for alvorlige bivirkninger fra legemidlet hos ammende spedbarn, bør det tas en beslutning om å avbryte ammingen eller å utsette eller avbryte behandlingen med REBETOL.
Pediatrisk bruk
Sikkerhet og effektivitet av REBETOL 200 mg i kombinasjon med PegIntron er ikke fastslått hos pediatriske pasienter under 3 år. For behandling med REBETOL/INTRON A, bør bevis på sykdomsprogresjon, som leverbetennelse og fibrose, samt prognostiske faktorer for respons, HCV-genotype og viral belastning vurderes når man bestemmer seg for å behandle en pediatrisk pasient. Fordelene med behandlingen bør veies opp mot sikkerhetsfunnene som er observert.
Langtidsoppfølgingsdata hos pediatriske personer indikerer at REBETOL i kombinasjon med PegIntron eller med INTRON A kan indusere en vekstinhibering som resulterer i redusert høyde hos noen pasienter [se ADVARSLER OG FORHOLDSREGLER og BIVIRKNINGER ].
Selvmordstanker eller selvmordsforsøk forekom hyppigere blant pediatriske pasienter, først og fremst ungdom, sammenlignet med voksne pasienter (2,4 % vs. 1 %) under behandling og oppfølging utenfor behandlingen [se ADVARSLER OG FORHOLDSREGLER ]. Som hos voksne pasienter opplevde pediatriske pasienter andre psykiatriske bivirkninger (f.eks. depresjon, emosjonell labilitet, somnolens), anemi og nøytropeni [se ADVARSLER OG FORHOLDSREGLER ].
Geriatrisk bruk
Kliniske studier med REBETOL/INTRON A- eller PegIntron-behandling inkluderte ikke tilstrekkelig antall forsøkspersoner i alderen 65 år og over til å avgjøre om de responderer annerledes enn yngre forsøkspersoner.
REBETOL 200mg er kjent for å utskilles vesentlig av nyrene, og risikoen for toksiske reaksjoner på dette legemidlet kan være større hos pasienter med nedsatt nyrefunksjon. Fordi eldre pasienter ofte har nedsatt nyrefunksjon, bør det utvises forsiktighet ved doseringsvalg. Nyrefunksjonen bør overvåkes og dosejusteringer bør gjøres tilsvarende. REBETOL 200mg skal ikke brukes til pasienter med kreatininclearance mindre enn 50 ml/min [se KONTRAINDIKASJONER ].
Generelt bør REBETOL 200 mg kapsler administreres til eldre pasienter med forsiktighet, med start i den nedre enden av doseringsområdet, noe som gjenspeiler den høyere frekvensen av nedsatt lever- og hjertefunksjon, og av samtidig sykdom eller annen medikamentell behandling. I kliniske studier hadde eldre forsøkspersoner en høyere frekvens av anemi (67 %) enn yngre pasienter (28 %) [se ADVARSLER OG FORHOLDSREGLER ].
Mottakere av organtransplantasjoner
Sikkerhet og effekt av INTRON A og PegIntron alene eller i kombinasjon med REBETOL for behandling av hepatitt C hos lever- eller andre organtransplanterte mottakere er ikke fastslått. I et lite (n=16) enkeltsenter, ukontrollert tilfelle, var nyresvikt hos nyre-allotransplanterte mottakere som fikk interferon alfa og ribavirin kombinasjonsbehandling hyppigere enn forventet fra senterets tidligere erfaring med nyre-allotransplanterte mottakere som ikke fikk kombinasjonsbehandling. Forholdet mellom nyresvikt og nyreallograftavstøtning er ikke klart.
HIV eller HBV co-infeksjon
Sikkerhet og effekt av PegIntron/REBETOL 200mg og INTRON A/REBETOL for behandling av pasienter med HCV samtidig-infisert med HIV eller HBV er ikke fastslått.
OVERDOSE
Det er begrenset erfaring med overdosering. Akutt inntak av opptil 20 g REBETOL-kapsler, INTRON A-inntak på opptil 120 millioner enheter og subkutane doser av INTRON A opptil 10 ganger anbefalte doser er rapportert. Primære effekter som er observert er økt forekomst og alvorlighetsgrad av bivirkningene relatert til terapeutisk bruk av INTRON A og REBETOL. Imidlertid har leverenzymavvik, nyresvikt, blødning og hjerteinfarkt blitt rapportert ved administrering av enkelt subkutane doser av INTRON A som overstiger doseringsanbefalingene.
Det finnes ingen spesifikk motgift for INTRON A eller REBETOL 200 mg overdose, og hemodialyse og peritonealdialyse er ikke effektive for behandling av overdose av disse midlene.
KONTRAINDIKASJONER
REBETOL kombinasjonsbehandling er kontraindisert ved:
- . kvinner som er gravide. REBETOL 200mg kan forårsake fosterskader når det gis til en gravid kvinne. REBETOL er kontraindisert hos kvinner som er eller kan bli gravide. Hvis REBETOL brukes under graviditet, eller hvis pasienten blir gravid mens hun tar REBETOL 200 mg, bør pasienten informeres om den potensielle faren for fosteret hennes [se ADVARSLER OG FORHOLDSREGLER , Bruk i spesifikke populasjoner , og PASIENTINFORMASJON ] . menn hvis kvinnelige partnere er gravide . pasienter med kjente overfølsomhetsreaksjoner som Stevens-Johnsons syndrom, toksisk, epidermal nekrolyse og erythema multiforme overfor ribavirin eller en hvilken som helst komponent i produktet . pasienter med autoimmun hepatitt . pasienter med hemoglobinopatier (f.eks. thalassemia major, sigdcelleanemi) . pasienter med kreatininclearance mindre enn 50 ml/min. [se Bruk i spesifikke populasjoner og KLINISK FARMAKOLOGI ] . Samtidig administrering av REBETOL og didanosin er kontraindisert fordi eksponering for den aktive metabolitten av didanosin (dideoksyadenosin 5'-trifosfat) er økt. Fatal leversvikt, så vel som perifer nevropati, pankreatitt og symptomatisk hyperlaktatemi/laktacidose er rapportert hos pasienter som får didanosin i kombinasjon med ribavirin [se NARKOTIKAHANDEL ].
KLINISK FARMAKOLOGI
Virkningsmekanismen
Ribavirin er et antiviralt middel [se Mikrobiologi ].
Farmakokinetikk
Enkelt- og flerdose farmakokinetiske egenskaper hos voksne er oppsummert i tabell 11. Ribavirin ble raskt og omfattende absorbert etter oral administrering. På grunn av first-pass metabolisme var imidlertid den absolutte biotilgjengeligheten i gjennomsnitt 64 % (44 %). Det var en lineær sammenheng mellom dose og AUCtf (AUC fra tid null til siste målbare konsentrasjon) etter enkeltdoser på 200 til 1200 mg ribavirin. Forholdet mellom dose og Cmax var krumlinjet, og hadde en tendens til asymptoter over enkeltdoser på 400 til 600 mg.
Ved gjentatt oral dosering, basert på AUC12t, ble det observert en 6 ganger akkumulering av ribavirin i plasma. Etter oral dosering med 600 mg to ganger daglig, ble steady-state nådd etter ca. 4 uker, med gjennomsnittlige steady-state plasmakonsentrasjoner på 2200 ng/ml (37%). Ved seponering av doseringen var gjennomsnittlig halveringstid 298 (30 %) timer, noe som sannsynligvis reflekterer langsom eliminasjon fra ikke-plasmakompartmenter.
Effekt av antacida på absorpsjon av ribavirin
Samtidig administrering av REBETOL-kapsler med et syrenøytraliserende middel som inneholder magnesium, aluminium og simetikon resulterte i en 14 % reduksjon i gjennomsnittlig AUCtf for ribavirin. Den kliniske relevansen til resultatene fra denne enkeltdosestudien er ukjent.
Vevsdistribusjon
Ribavirintransport inn i ikke-plasma-rom har blitt mest omfattende studert i røde blodlegemer, og har blitt identifisert å være primært via en ekvilibrativ nukleosidtransportør av es-type. Denne typen transportører finnes på praktisk talt alle celletyper og kan stå for det omfattende distribusjonsvolumet. Ribavirin binder seg ikke til plasmaproteiner.
Metabolisme og utskillelse
Ribavirin har to metabolismeveier: (i) en reversibel fosforyleringsvei i kjerneholdige celler; og (ii) en nedbrytningsvei som involverer deribosylering og amidhydrolyse for å gi en triazolkarboksylsyremetabolitt. Ribavirin og dets triazolkarboksamid- og triazolkarboksylsyremetabolitter utskilles renalt. Etter oral administrering av 600 mg 14C-ribavirin ble ca. 61 % og 12 % av radioaktiviteten eliminert i henholdsvis urin og feces i løpet av 336 timer. Uendret ribavirin utgjorde 17 % av den administrerte dosen.
Spesielle populasjoner
Nyredysfunksjon
Farmakokinetikken til ribavirin ble vurdert etter administrering av en enkelt oral dose (400 mg) ribavirin til ikke-HCV-infiserte personer med varierende grader av nyresvikt. Gjennomsnittlig AUCtf-verdi var tre ganger høyere hos personer med kreatininclearance-verdier mellom 10 og 30 ml/min sammenlignet med kontrollpersoner (kreatininclearance større enn 90 ml/min). Hos personer med kreatininclearance-verdier mellom 30 og 60 ml/min, var AUCtf dobbelt så høy sammenlignet med kontrollpersoner. Den økte AUCtf ser ut til å skyldes reduksjon av renal og nonrenal clearance hos disse pasientene. Fase 3-effektstudier inkluderte forsøkspersoner med kreatininclearance-verdier høyere enn 50 ml/min. Multidose-farmakokinetikken til ribavirin kan ikke forutsies nøyaktig hos pasienter med nedsatt nyrefunksjon. Ribavirin fjernes ikke effektivt ved hemodialyse.
Pasienter med kreatininclearance mindre enn 50 ml/min skal ikke behandles med REBETOL [se KONTRAINDIKASJONER ].
Leverdysfunksjon
Effekten av leverdysfunksjon ble vurdert etter en enkelt oral dose ribavirin (600 mg). Gjennomsnittlige AUCtf-verdier var ikke signifikant forskjellig hos personer med mild, moderat eller alvorlig leverdysfunksjon (Child-Pugh-klassifisering A, B eller C) sammenlignet med kontrollpersoner. Imidlertid økte gjennomsnittlige Cmax-verdier med alvorlighetsgraden av leverdysfunksjon og var dobbelt så høy hos personer med alvorlig leverdysfunksjon sammenlignet med kontrollpersoner.
Eldre pasienter
Farmakokinetiske evalueringer hos eldre personer er ikke utført.
Kjønn
Det var ingen klinisk signifikante farmakokinetiske forskjeller observert i en enkeltdosestudie med 18 mannlige og 18 kvinnelige forsøkspersoner.
Pediatriske pasienter
Multiple-dose farmakokinetiske egenskaper for REBETOL 200 mg kapsler og INTRON A hos pediatriske personer med kronisk hepatitt C mellom 5 og 16 år er oppsummert i tabell 12. Farmakokinetikken til REBETOL 200 mg og INTRON A (dosenormaliserte og) er lik hos voksne pediatriske fag.
Fullstendige farmakokinetiske egenskaper til REBETOL mikstur er ikke bestemt hos pediatriske personer. Ribavirin Cmin-verdier var like etter administrering av REBETOL mikstur eller REBETOL 200 mg kapsler i løpet av 48 ukers behandling hos pediatriske personer (3 til 16 år).
En klinisk studie med pediatriske personer med kronisk hepatitt C mellom 3 og 17 år ble utført der farmakokinetikken for PegIntron og REBETOL (kapsler og mikstur) ble evaluert. Hos pediatriske forsøkspersoner som fikk kroppsoverflate-justert dosering av PegIntron på 60 mcg/m²/uke, ble estimatet for log transformert ratio av eksponering under doseringsintervallet spådd å være 58 % [90 % KI: 141 %, 177 %] høyere enn observert hos voksne som får 1,5 mikrogram/kg/uke. Farmakokinetikken til REBETOL (dosenormalisert) i denne studien var lik den som ble rapportert i en tidligere studie med REBETOL 200 mg i kombinasjon med INTRON A hos pediatriske personer og voksne.
Effekt av mat på absorpsjon av ribavirin
Både AUCtf og Cmax økte med 70 % når REBETOL-kapsler ble administrert sammen med et fettrikt måltid (841 kcal, 53,8 g fett, 31,6 g protein og 57,4 g karbohydrat) i en enkeltdose farmakokinetisk studie [se DOSERING OG ADMINISTRASJON ].
Mikrobiologi
Virkningsmekanismen
Mekanismen som ribavirin bidrar til sin antivirale effekt i klinikken er ikke fullt ut forstått. Ribavirin har direkte antiviral aktivitet i vevskultur mot mange RNA-virus. Ribavirin øker mutasjonsfrekvensen i genomene til flere virus og ribavirintrifosfat hemmer HCV-polymerase i en biokjemisk reaksjon.
Antiviral aktivitet i cellekultur
Den antivirale aktiviteten til ribavirin i HCV-replikonet er ikke godt forstått og har ikke blitt definert på grunn av den cellulære toksisiteten til ribavirin. Direkte antiviral aktivitet er observert i vevskultur av andre RNA-virus. Anti-HCV-aktiviteten til interferon ble demonstrert i celle som inneholdt selvreplikerende HCV-RNS (HCV-replikonceller) eller HCV-infeksjon.
Motstand
HCV-genotyper viser stor variasjon i deres respons på pegylert rekombinant human interferon/ribavirinbehandling. Genetiske endringer assosiert med variabel respons er ikke identifisert.
Kryssmotstand
Det er ingen rapportert kryssresistens mellom pegylerte/ikke-pegylerte interferoner og ribavirin.
Dyretoksikologi og farmakologi
Langtidsstudier på mus og rotter [18 til 24 måneder; doser på henholdsvis 20 til 75 og 10 til 40 mg/kg/dag (estimerte humanekvivalente doser på henholdsvis 1,67 til 6,25 og 1,43 til 5,71 mg/kg/dag, basert på kroppsoverflatejustering for en voksen på 60 kg; ca. 0,1 til 0,4 ganger den maksimale humane 24-timers dosen av ribavirin)] har vist en sammenheng mellom kronisk ribavirineksponering og økt forekomst av vaskulære lesjoner (mikroskopiske blødninger) hos mus. Hos rotter forekom netthinnedegenerasjon hos kontroller, men forekomsten var økt hos ribavirinbehandlede rotter.
en studie der rotteunger ble dosert postnatalt med ribavirin i doser på 10, 25 og 50 mg/kg/dag, skjedde legemiddelrelaterte dødsfall ved 50 mg/kg (ved plasmakonsentrasjoner av rotteunger under plasmakonsentrasjoner hos mennesker hos mennesker) terapeutisk dose) mellom studiedag 13 og 48. Rotteunger dosert fra postnatale dag 7 til og med 63 viste en mindre, doserelatert reduksjon i total vekst ved alle doser, som deretter ble manifestert som en liten reduksjon i kroppsvekt, krone-rumpelengde, og beinlengde. Disse effektene viste tegn på reversibilitet, og ingen histopatologiske effekter på bein ble observert. Ingen ribavirineffekter ble observert angående nevroadferdsmessig eller reproduktiv utvikling.
Kliniske studier
Klinisk studie 1 evaluerte PegIntron monoterapi. Se PegIntron-merking for informasjon om denne studien.
REBETOL/PegIntron kombinasjonsterapi
Voksne emner
Studie 2
En randomisert studie sammenlignet behandling med to PegIntron/REBETOL 200 mg-regimer [PegIntron 1,5 mcg/kg subkutant én gang ukentlig/REBETOL 800 mg oralt daglig (i oppdelte doser); PegIntron 1,5 mcg/kg subkutant én gang ukentlig i 4 uker, deretter 0,5 mcg/kg subkutant én gang ukentlig i 44 uker/REBETOL 1000 eller 1200 mg oralt daglig (i oppdelte doser)] med INTRON A [3 MIE subkutant tre ganger ukentlig/REBET eller REBET 1200 mg oralt daglig (i oppdelte doser)] hos 1530 voksne med kronisk hepatitt C. Interferon-naive forsøkspersoner ble behandlet i 48 uker og fulgt i 24 uker etter behandling. Kvalifiserte personer hadde kompensert leversykdom, påviselig HCV-RNA, forhøyet ALAT og leverhistopatologi i samsvar med kronisk hepatitt.
Respons på behandling ble definert som upåviselig HCV-RNA 24 uker etter behandling (se tabell 13). Responsraten på dosen PegIntron 1,5 mcg/kg og ribavirin 800 mg var høyere enn responsraten på INTRON A/REBETOL (se tabell 13). Responsraten på PegIntron 1,5→0,5 mcg/kg/REBETOL 200 mg var i hovedsak den samme som svar på INTRON A/REBETOL (data ikke vist).
Personer med viral genotype 1, uavhengig av viral mengde, hadde en lavere responsrate på PegIntron (1,5 mcg/kg)/REBETOL (800 mg) sammenlignet med forsøkspersoner med andre virale genotyper. Personer med både dårlige prognostiske faktorer (genotype 1 og høy viral mengde) hadde en responsrate på 30 % (78/256) sammenlignet med en responsrate på 29 % (71/247) med INTRON A/REBETOL kombinasjonsbehandling.
Personer med lavere kroppsvekt hadde en tendens til å ha høyere bivirkningsfrekvens [se BIVIRKNINGER og høyere responsrate enn forsøkspersoner med høyere kroppsvekt. Forskjeller i responsrater mellom behandlingsarmene varierte ikke vesentlig med kroppsvekt.
Behandlingsresponsrater med PegIntron/REBETOL kombinasjonsbehandling var 49 % hos menn og 56 % hos kvinner. Responsratene var lavere hos afroamerikanske og latinamerikanske personer og høyere hos asiater sammenlignet med kaukasiere. Selv om afroamerikanere hadde en høyere andel dårlige prognostiske faktorer sammenlignet med kaukasiere, var antallet ikke-kaukasiere studert (11 % av totalen) utilstrekkelig til å tillate meningsfulle konklusjoner om forskjeller i responsrater etter justering for prognostiske faktorer i denne studien.
Leverbiopsier ble tatt før og etter behandling hos 68 % av pasientene. Sammenlignet med baseline ble omtrent to tredjedeler av pasientene i alle behandlingsgruppene observert å ha en beskjeden reduksjon i betennelse.
Studie 3
en stor samfunnsbasert studie i USA ble 4913 personer med kronisk hepatitt C randomisert til å få PegIntron 1,5 mcg/kg subkutant én gang ukentlig i kombinasjon med en REBETOL 200 mg dose på 800 til 1400 mg (vektbasert dosering [WBD]) eller 800 mg (flat) oralt daglig (i oppdelte doser) i 24 eller 48 uker basert på genotype. Respons på behandling ble definert som upåviselig HCV-RNA (basert på en analyse med en nedre deteksjonsgrense på 125 IE/mL) 24 uker etter behandling.
Behandling med PegIntron 1,5 mcg/kg og REBETOL 800 til 1400 mg resulterte i en høyere vedvarende virologisk respons sammenlignet med PegIntron i kombinasjon med en flat 800 mg daglig dose av REBETOL. Personer som veier mer enn 105 kg oppnådde størst fordel med WBD, selv om en beskjeden fordel også ble observert hos personer som veide over 85 til 105 kg (se tabell 14). Fordelen med WBD hos forsøkspersoner som veier mer enn 85 kg ble observert med HCV genotype 1-3. Det var utilstrekkelig data tilgjengelig til å trekke konklusjoner angående andre genotyper. Bruk av WBD resulterte i økt forekomst av anemi [se BIVIRKNINGER ].
Totalt 1552 personer som veide mer enn 65 kg i studie 3 hadde genotype 2 eller 3 og ble randomisert til 24 eller 48 ukers behandling. Ingen ytterligere fordel ble observert med lengre behandlingsvarighet.
Studie 4
En stor randomisert studie sammenlignet sikkerhet og effekt av behandling i 48 uker med to PegIntron/REBETOL 200 mg-regimer [PegIntron 1,5 mcg/kg og 1 mcg/kg subkutant en gang ukentlig, begge i kombinasjon med REBETOL 800 til 1400 mg PO daglig (i to delt) doser)] og Pegasys 180 mcg subkutant én gang ukentlig i kombinasjon med Copegus 1000 til 1200 mg PO daglig (i to oppdelte doser) hos 3070 behandlingsnaive voksne med kronisk hepatitt C genotype 1. I denne studien, mangel på tidlig virologisk respons (upåviselig) HCV-RNA eller større enn eller lik 2 log10 reduksjon fra baseline) ved behandling Uke 12 var kriteriet for seponering av behandlingen. SVR ble definert som upåviselig HCV-RNA (Roche COBAS TaqMan-analyse, en nedre grense for kvantifisering på 27 IE/ml) 24 uker etter behandling (se tabell 15).
Samlet SVR-frekvens var lik blant de tre behandlingsgruppene. Uavhengig av behandlingsgruppe var SVR-frekvensene lavere hos personer med dårlige prognostiske faktorer. Personer med dårlige prognostiske faktorer randomisert til PegIntron (1,5 mcg/kg)/REBETOL eller Pegasys/Copegus, oppnådde imidlertid høyere SVR-frekvenser sammenlignet med lignende forsøkspersoner randomisert til PegIntron 1 mcg/kg/REBETOL. For dosen PegIntron 1,5 mcg/kg og REBETOL var SVR-rater for forsøkspersoner med og uten følgende prognostiske faktorer som følger: skrumplever (10 % vs. 42 %), normale ALAT-nivåer (32 % vs. 42 %), virus ved baseline belastning større enn 600 000 IE/ml (35 % vs. 61 %), 40 år og eldre (38 % vs. 50 %) og afroamerikansk rase (23 % vs. 44 %). Hos personer med upåviselig HCV-RNA ved behandlingsuke 12 som fikk PegIntron (1,5 mcg/kg)/REBETOL 200 mg, var SVR-raten 81 % (328/407).
Studie 5 - REBETOL/PegIntron kombinasjonsterapi ved tidligere behandlingssvikt
en ikke-komparativ studie ble 2293 personer med moderat til alvorlig fibrose som mislyktes med tidligere behandling med kombinasjon alfa-interferon/ribavirin, behandlet på nytt med PegIntron, 1,5 mikrogram/kg subkutant, en gang ukentlig, i kombinasjon med vektjustert ribavirin. Kvalifiserte forsøkspersoner inkluderte tidligere ikke-respondere (pasienter som var HCV-RNA-positive ved slutten av minimum 12 ukers behandling) og tidligere tilbakefallende (pasienter som var HCVRNA-negative ved slutten av minimum 12 ukers behandling og deretter fikk tilbakefall etter behandling etter behandling). følge opp). Personer som var negative i uke 12 ble behandlet i 48 uker og fulgt i 24 uker etter behandling. Respons på behandling ble definert som upåviselig HCV-RNA 24 uker etter behandling (målt ved hjelp av en forskningsbasert test, deteksjonsgrense 125 IE/ml). Den samlede svarprosenten var 22 % (497/2293) (99 % KI: 19,5, 23,9). Pasienter med følgende egenskaper hadde mindre sannsynlighet for å dra nytte av ny behandling: tidligere ikke-respons, tidligere pegylert interferonbehandling, signifikant brodannende fibrose eller skrumplever og genotype 1-infeksjon.
De vedvarende virologiske responsratene ved gjenbehandling etter baseline-karakteristikker er oppsummert i tabell 16.
Oppnåelse av et upåviselig HCV-RNA ved behandlingsuke 12 var en sterk prediktor for SVR. I denne studien oppnådde ikke 1470 (64 %) forsøkspersoner et upåviselig HCV-RNA ved behandlingsuke 12, og ble tilbudt å delta i langtidsbehandlingsstudier på grunn av utilstrekkelig behandlingsrespons. Av de 823 (36 %) forsøkspersonene som ikke kunne påvises HCV-RNA ved behandlingsuke 12, hadde de infisert med genotype 1 en SVR på 48 % (245/507), med en rekke responser etter fibroseskåre (F4-F2) på 39-55 %. Forsøkspersoner infisert med genotype 2/3 som var HCV-RNA upåviselig ved behandlingsuke 12 hadde en total SVR på 70 % (196/281), med en rekke responser etter fibrose-skåre (F4-F2) på 60-83 %. For alle genotyper var høyere fibrosescore assosiert med en redusert sannsynlighet for å oppnå SVR.
Pediatriske fag
Tidligere ubehandlede pediatriske forsøkspersoner i alderen 3 til 17 år med kompensert kronisk hepatitt C og detekterbart HCV-RNA ble behandlet med REBETOL 15 mg/kg per dag og PegIntron 60 mcg/m² én gang ukentlig i 24 eller 48 uker basert på HCV-genotype og baseline viral. laste. Alle forsøkspersoner skulle følges i 24 uker etter behandling. Totalt 107 forsøkspersoner mottok behandling, hvorav 52 % var kvinner, 89 % var kaukasiske og 67 % var infisert med HCV genotype 1. Personer infisert med genotype 1, 4 eller genotype 3 med HCV-RNA større enn eller lik 600 000 IE/ml fikk 48 ukers behandling mens de som var infisert med genotype 2 eller genotype 3 med HCV-RNA mindre enn 600 000 IE/ml fikk 24 ukers behandling. Prøveresultatene er oppsummert i tabell 17.
REBETOL/INTRON En kombinasjonsterapi
Voksne emner
Tidligere ubehandlede personer
Voksne med kompensert kronisk hepatitt C og påviselig HCV-RNA (vurdert av et sentralt laboratorium ved bruk av en forskningsbasert RT-PCR-analyse) som tidligere var ubehandlet med alfa-interferonterapi, ble registrert i to multisenter, dobbeltblindede studier (USA og internasjonale) og randomisert til å motta REBETOL 200 mg kapsler 1200 mg/dag (1000 mg/dag for personer som veier mindre enn eller lik 75 kg) og INTRON A 3 MIE tre ganger ukentlig eller INTRON A og placebo i 24 eller 48 uker etterfulgt av 24 uker med oppfølging utenfor terapi. Den internasjonale studien inneholdt ikke en 24-ukers INTRON A- og placebobehandlingsarm. Den amerikanske studien inkluderte 912 forsøkspersoner som ved baseline var 67 % menn, 89 % kaukasiske med en gjennomsnittlig Knodell HAI-score (I+II+III) på 7,5 og 72 % genotype 1. Den internasjonale studien, utført i Europa, Israel , Canada og Australia, registrerte 799 personer (65 % menn, 95 % kaukasiske, gjennomsnittlig Knodell-score 6,8 og 58 % genotype 1).
Prøveresultatene er oppsummert i tabell 18.
Av forsøkspersoner som ikke hadde oppnådd HCV-RNA under deteksjonsgrensen for den forskningsbaserte analysen innen uke 24 av REBETOL/INTRON A-behandling, svarte mindre enn 5 % på ytterligere 24 ukers kombinasjonsbehandling.
Blant forsøkspersoner med HCV genotype 1 behandlet med REBETOL/INTRON A-terapi som oppnådde HCV-RNA under deteksjonsgrensen for den forskningsbaserte analysen med 24 uker, hadde de randomisert til 48 ukers behandling høyere virologisk respons sammenlignet med de i 24- ukes behandlingsgruppe. Det ble ikke observert økning i responsrater for personer med HCV ikke-genotype 1 randomisert til REBETOL/INTRON A-behandling i 48 uker sammenlignet med 24 uker.
Tilbakefallsfag
Pasienter med kompensert kronisk hepatitt C og påviselig HCV-RNA (vurdert av et sentrallaboratorium ved bruk av en forskningsbasert RT-PCR-analyse) som hadde fått tilbakefall etter en eller to kurer med interferonbehandling (definert som unormale serum-ALAT-nivåer) ble registrert i to multisenter, dobbeltblindede studier (USA og internasjonale) og randomisert til å motta REBETOL 1200 mg/dag (1000 mg/dag for forsøkspersoner som veier ≤ 75 kg) og INTRON A 3 MIE tre ganger ukentlig eller INTRON A og placebo i 24 uker etterfulgt av 24 uker med oppfølging utenom terapi. Den amerikanske studien inkluderte 153 forsøkspersoner som ved baseline var 67 % menn, 92 % kaukasiske med en gjennomsnittlig Knodell HAI-score (I+II+III) på 6,8 og 58 % genotype 1. Den internasjonale studien, utført i Europa, Israel , Canada og Australia, registrerte 192 forsøkspersoner (64 % menn, 95 % kaukasiske, gjennomsnittlig Knodell-score 6,6 og 56 % genotype 1). Prøveresultatene er oppsummert i tabell 19.
Virologiske og histologiske responser var like blant mannlige og kvinnelige forsøkspersoner i både de tidligere ubehandlede og tilbakefallsforsøkene.
Pediatriske fag
Pediatriske personer i alderen 3 til 16 år med kompensert kronisk hepatitt C og påviselig HCV-RNA (vurdert av et sentrallaboratorium ved bruk av en forskningsbasert RT-PCR-analyse) ble behandlet med REBETOL 15 mg/kg per dag og INTRON A 3 MIE/ m² tre ganger ukentlig i 48 uker etterfulgt av 24 ukers oppfølging uten behandling. Totalt 118 personer mottok behandling, hvorav 57 % var menn, 80 % kaukasiske og 78 % genotype 1. Personer under 5 år fikk REBETOL mikstur, og de som var 5 år eller eldre fikk enten REBETOL mikstur eller eldre. kapsler.
Prøveresultatene er oppsummert i tabell 20.
Personer med viral genotype 1, uavhengig av viral belastning, hadde en lavere responsrate på INTRON A/REBETOL kombinasjonsbehandling sammenlignet med forsøkspersoner med genotype non-1, 36 % vs. 81 %. Personer med både dårlige prognostiske faktorer (genotype 1 og høy virusmengde) hadde en responsrate på 26 % (13/50).
PASIENTINFORMASJON
Ingen informasjon gitt. Vennligst referer til ADVARSLER OG FORHOLDSREGLER seksjon.